2. Mitochondria and NAFLD
Mitochondria are highly dynamic organelles that serve as the center for energy metabolism. The liver consumes about 15% of the organism’s oxygen under normal conditions, implying that hepatocytes are rich in mitochondria, which require oxygen to make adenosine triphosphate (ATP) [
93]. Mitochondria make up to 18% of the total volume of a hepatocytes and play a critical role in the liver’s metabolic functions as well as nutrient (carbohydrates, lipids, and proteins) oxidation for energy generation [
94]. The balance between mitochondrial biogenesis, mitochondrial fission/fusion, and mitochondrial autophagy is essential for mitochondrial homeostasis. Emerging evidence suggests that cellular functions are at risk when mitochondrial homeostasis is disrupted [
95,
96].
2.1. Mitochondrial Fatty Acid Oxidation
Mitochondria are the cells’ “powerhouse”, as they are the primary source of energy. Inside mitochondria, dietary glucose and fatty acids are oxidized through β-oxidation and the tricarboxylic acid (TCA) cycle. In fatty acid catabolism, FFAs are converted to fatty acyl-CoA in the hepatocyte cytosol, which then enters into mitochondria via carnitine palmitoyl-transferase 1 (CPT1). The first step of β-oxidation of long-chain fatty acids is the dehydrogenation of the acyl-CoA easter by acyl-CoA dehydrogenases followed by the three steps carried out by mitochondrial trifunctional protein (MTP). The β-oxidation breaks the FFA into acetyl CoA, which can be degraded to CO2 and H2O by the TCA cycle. The electron transport chain (ETC), which is found in the inner mitochondrial membrane, is the main site of ATP production. It is made up of five complexes numbered from I to V. This aerobic process necessitates the presence of a reducing component, which is provided by the electron carriers NADH and FADH2. Electron transfer occurs from complex I to complex IV by providing energy to pump protons from the mitochondrial matrix to the intermembranous region, which will lead to a proton gradient. The potential energy in this gradient is then used by complex V for the synthesis of ATP.
2.2. Mitochondrial Dynamics, Biogenesis and Mitophagy
Mitochondrial dynamics involve the fusion and fission of mitochondria [
97]. Under the conditions of metabolic or environmental stress, mitochondrial fission and fusion processes are critical for maintaining the function of the mitochondria. Fusion is a process in which the contents of partially damaged mitochondria are mixed to promote complementation, which helps to mitigate cellular stress. Fission is a process that leads to the creation of new mitochondria, which also contributes to quality control by separating the damaged mitochondria from the healthy mitochondria [
98,
99]. Disruptions of these processes have been implicated in disease. In mammalian cells, mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2) are responsible for the fusion process of the outer mitochondrial membrane [
100], while the optic atrophy 1 (Opa1) protein regulates the fusion of the inner mitochondrial membrane [
101,
102]. The primary proteins involved in fission are the dynamin-related protein 1 (Drp1) and Mitochondrial Fission 1 Protein (Fis1) [
103,
104]. In mitochondrial bioenergetics, the balance between fusion and fission processes is critical. Intracellular stress and external factors can disrupt the balance between fusion and fission, resulting in mitochondrial fragmentation [
105]. Excessive fission is characterized by increased levels of the fission protein dynamin-related protein 1 (Drp1). Mitochondrial dysfunction is associated with the dysregulation of the proteins involved in mitochondrial fission [
106,
107].
Mitochondrial biogenesis comprises mitochondrial DNA (mtDNA) replication, the transcription of mtDNA and nuclear coding genes, and the translation and assembly of the OXPHOS complex, all of which are important for cellular homeostasis and survival [
108]. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1α) is a master regulator of mitochondrial biogenesis and a major transcriptional activator [
109]. It regulates mitochondrial biogenesis by regulating the expression of nuclear and mitochondrial genes by activating a number of other transcription factors such as nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2), resulting in the expression of mitochondrial transcription factor A (TFAM) [
110,
111]. TFAM interacts directly with the mitochondrial genome and mitochondrial transcription factors, resulting in the transcription of mitochondrial genes [
112]. Furthermore, PGC-1α increases mitochondrial FAO by acting as a co-activator of peroxisome proliferator-activated receptor α and γ (PPARα and PPARγ), which leads to the production of mitochondrial β-oxidation genes [
113,
114]. Moreover, PGC-1α activation stimulates mitochondrial biogenesis as manifested by the stimulation of mitochondrial DNA replication and mitochondria gene expression [
115,
116]. Through the phosphorylation of PGC-1α, AMP-activated protein kinase (AMPK) is also involved in the regulation of mitochondrial biogenesis [
117].
Mitophagy is a mitochondrial-specific autophagy that involves the selective isolation and degradation of damaged mitochondria to maintain the functional integrity and cellular homeostasis [
118]. The PTEN-induced kinase 1 (PINK1)—Parkin pathway, plays an important role during mitophagy [
119]. Mitophagy is a protective mechanism enabling the cell to avoid generating reactive oxygen species (ROS) due to damaged mitochondria and maintains redox balance [
120]. In addition, mitophagy also stimulates lipid droplet breakdown and the release of FFAs, which are then transported to healthy mitochondria for β-oxidation and increased energy release.
2.3. Role of Mitochondrial Dysfunction in NAFLD
Mitochondrial dysfunction and oxidative stress have been observed in liver tissue from patients with fatty liver disease [
121]. It is characterized by various degrees of ultrastructural mitochondrial damage, abnormal morphologic changes, respiratory chain activity reduction, ATP depletion, increased permeability of the outer and inner membranes, ROS overproduction, oxidative stress-mediated deletions of mtDNA, and impaired mitochondrial β-oxidation [
50,
122]. Several recent investigations have suggested that mitochondrial dysfunction is a major factor in the development and progression of NAFLD [
123]. The exact mechanism by which mitochondrial dysfunction contributes to NAFLD is still not fully understood. Reduced FAO, increased FFA delivery and transport into the liver, and increased hepatic fatty acid synthesis have been reported to be involved in the pathogenesis of NAFLD [
121].
2.3.1. FAO and NAFLD
Mitochondrial dysfunction has been linked to a reduction in the β-oxidation of lipids, resulting in triglyceride accumulation in hepatocytes [
124]. In humans with obesity and NAFLD, a moderately malfunctioning hepatocyte ETC has been reported [
51]. High amounts of malonyl-CoA have been shown to inhibit CPT-1, leading to a reduction in β-oxidation [
121]. Acylcarnitine accumulation has also been reported as a marker of mitochondrial stress, malfunction, and reduced FAO [
125]. Impaired β-oxidation influences peroxisomal and cytochrome oxidation of FFAs, resulting in significant levels of ROS and hazardous byproducts [
54]. Our group has previously reported that defects in mitochondrial FAO induce hepatic steatosis in mice without progressing to cirrhosis [
126,
127]. Human MTP defects are recessively inherited, and children who are deficient in one or more of the three enzymatic functions in MTP have been found to have microvesicular hepatic steatosis [
128]. Our previous research has shown that homozygous mice with MTP deficiency develop hepatic steatosis shortly after birth [
129]. Further, we previously reported that aged mice heterozygous for MTP abnormalities develop insulin resistance and hepatic steatosis [
126]. Finally, we observed that MTP modulation rescued NAFLD in mice [
130].
More recently, our group has investigated the relationship between NAFLD/NASH, mitochondrial fatty acid oxidation in liver, and hepatic mitochondrial quality in human subjects [
131]. We obtained liver biopsies from patients with obesity undergoing bariatric surgery and correlated the liver histology to the mitochondrial fatty acid oxidation measured in the liver tissue. The results of this study showed that hepatic mitochondrial complete FAO was reduced by ~40–50% in subjects with NASH compared to the control subjects with normal histology. This decrease corresponded with increased hepatic mitochondrial reactive oxygen species production and reductions in the markers of mitochondrial biogenesis and mitophagy [
131]. These findings support a link between mitochondrial dysfunction and NAFLD/NASH in humans and suggest that impaired hepatic fatty acid oxidation and reduced mitochondrial quality are strongly linked to increasing NAFLD severity in patients with obesity. Further, in our ongoing study in veterans undergoing liver biopsy for suspected NASH, measurement of the FAO in the liver tissue showed that hepatic FAO was ~40% lower in veterans with liver fibrosis compared to those without fibrosis (unpublished data recently presented in abstract format at the American Association for the Study of Liver Diseases (AASLD) annual meeting [
132]). There were no significant differences in the FAO among various fibrosis stages. These findings suggest that compromised hepatic mitochondrial fatty acid oxidation is linked to NAFLD progression and fibrosis development in the veteran population. Our human studies are the first to directly measure mitochondrial FAO and mitochondrial quality in liver tissue obtained from NAFLD/NASH patients.
Mitochondrial dysfunction results from oxidative mitochondrial damage, with abnormalities in the mitochondrial ETC and OXPHOS [
133]. NAFLD patients have been found to have a higher rate of mtDNA mutations, including genes encoding ETC complexes, with the mutational burden increasing with the severity of the histopathological abnormalities [
134]. Reduced expression of their corresponding messenger RNA was linked to mutations in genes encoding ETC subunits [
134]. Previous studies have reported that in the liver tissue of patients and animal models with NAFLD, the mitochondrial respiratory chain complex is reduced [
125,
135]. Compared to individuals without NAFLD, patients with NAFLD have been found to have a reduction in respiratory chain activity of 37% in complex I, 42% in complex II, 30% in complex III, 38% in complex IV, and 58% in complex V [
125]. According to a previous report, complex I and IV subunits were drastically reduced in CDD-fed mice [
136].
2.3.2. Insulin Resistance and NAFLD
Insulin resistance (IR) has been linked to reduced mitochondrial numbers, abnormal mitochondrial morphology, decreased levels of mitochondrial oxidative enzymes, and decreased ATP production both in vivo [
137,
138] and ex vivo in human muscle biopsies [
139]. Increased intracellular lipid levels caused by elevated FFA levels in the plasma are linked to insulin resistance in the liver and muscle [
140]. In adipocytes from type 2 diabetic patients or morbidly obese human beings, there are significantly less mitochondria, and fewer genes that are expressed during mitochondrial biogenesis [
141]. Hence, IR metabolic tissues, such as skeletal muscle, liver, and fat, frequently exhibit a decreased mitochondrial number, decreased mitochondrial gene expression, abnormal mitochondrial morphology, and abnormal activities in oxidative phosphorylation. These mitochondrial abnormalities are linked to insulin resistance, intracellular lipid synthesis, and the pathogenesis of type 2 diabetes and NAFLD. Finally, insulin resistance is caused by mitochondrial dysfunction, which increases ROS levels. These elevated ROS activate many serine kinases, which phosphorylate IRS proteins [
142]. As a result, increased ROS production brought on by lipid-induced mitochondrial dysfunction reduces insulin signaling both directly and indirectly. The angiopoietin-like protein 4 (ANGPTL4), which is released by the liver and adipose tissue [
143], is crucial for controlling the metabolism of lipids. Although ANGPTL4 has been linked to lipid homeostasis [
144], there is still debate on how it affects glucose metabolism [
145]. ANGPTL4 was found to promote lipolysis [
146] and to prevent the removal of triglycerides from plasma by inhibiting lipoprotein lipase [
147]. According to recent studies, mice who completed treadmill exercise also showed increased ANGPTL4 mRNA expression in the liver [
148].
In IR individuals and animal models, exercise increases insulin action and glucose tolerance [
149]. Significant evidence suggests that aerobic exercise promotes mitochondrial biogenesis by elevating PGC-1α, NRF-1, and TFAM gene expression. In order to improve whole-body glucose metabolism, endurance exercise training increases mitochondrial size, number, and oxidative activity [
150]. Skeletal muscle electron transport chain activity and mitochondrial cristae are increased by moderate-intensity exercise and weight loss, improving insulin sensitivity [
151]. Aerobic exercise restores the age-related decline in mitochondrial gene expression and mitochondrial biogenesis [
152]. Exercise therefore enhances glucose and lipid metabolism by AMPK and PGC-1 activation, which increases mitochondrial biogenesis and function.
2.3.3. ROS and NAFLD
Excess ROS production in NAFLD has been reported to be linked to ETC disruption, permeabilization of the outer mitochondrial membrane, altered mitochondrial membrane potential (Δ
Ψm), and alterations in mitochondrial structural integrity [
52]. Excess ROS has been shown to induce oxidative base lesions in mtDNA bases, which has been shown to induce apoptosis [
153,
154,
155]. Mitochondrial DNA mutations caused by oxidative modifications are damaging to cellular integrity and are linked to higher ROS leakage from organelles and cells [
156]. The initial cause of this ROS-damaging cycle is believed to be long-chain FFAs, which accumulate in nonalcoholic fatty liver [
157]. It has been previously reported that NAFLD is characterized by mtDNA depletion and elevated liver levels of 8-hydroxy-2’-deoxyguanosine (8-OHdG), a DNA oxidation marker [
51]. We previously observed that ultrastructural abnormalities in the mitochondria of an NAFLD rat model, such as cristae disruption, hypodense matrix, and swelling/rounding, were strongly associated with enhanced ROS generation [
52]. When ROS stimulates NF-κB and the nuclear-binding oligomerization domain-like receptor family and the pyrin domain-containing 3 (NLRP3) inflammasome, inflammatory cytokines such as IL-1β, IL-6, and TNF-α are generated [
158]. Mitochondrial permeability transition pores (MPTP), which release mtDNA into the cytoplasm and serve as a danger-associated molecular pattern (DAMP), can arise as a result of mitochondrial membrane disruption caused by ROS. The NLRP3 inflammasome is subsequently activated by MPTP, which causes the cytokine IL-1β to mature and prolong inflammation [
159]. These proinflammatory cytokines also activate Kupffer cells, which produce proinflammatory cytokines that increase the inflammatory response in the liver. As a result of MPTP synthesis, immunological mediator cells such as Toll-like receptor 9 (TLR9) and formyl peptide receptor 1 (FPR1) are activated, leading to hepatocyte necrosis and the extracellular release of mitochondrial substances such as mitochondrial DNA and N-formyl peptide [
160,
161,
162].
In the livers of rodents fed HFD or choline-deficient diets or an ethionine-supplemented (CDE) diet, PGC-1α and mitochondrial biogenesis were suppressed along with TFAM downregulation [
163,
164]. Sirt1 overexpression in the nucleus and Sirt3 overexpression in the mitochondria have been demonstrated to improve antioxidant defense enzymes and decrease proinflammatory pathways [
165], providing protection against NAFLD and acting as sensors to promote PGC-1α expression. Sirt3 has been found to inhibit the adaptive response to high levels of hepatic FFA, resulting in mitochondrial dysfunction [
166,
167]. As a result, in the early stages of hepatocyte steatosis, primary mitochondrial dysfunction activates multiple adaptive metabolic pathways to reduce oxidative damage, which are mediated by increased mitochondrial activity and the increased expression of ROS detoxification genes by PGC-1α as well as Sirt1 and Sirt3 [
168]. The PGC-α1-NRF-2 complex causes mitochondrial transcription factors B1 and B2 (tfb1m and tfb2m) to be expressed in mice [
169]. Further, mitophagy is increased, and mitochondrial mass as well as mtDNA and PGC-1α expression are decreased in advanced NAFLD, all of which contribute to a vicious cycle of hepatic mitochondrial depletion and dysfunction. Deficient regulation of hepatocyte mitophagy promotes enhanced oxidative stress and inflammation in the late versus early stages of NAFLD [
170,
171].
Patients with NASH have been found to have ultrastructurally damaged mitochondria in their liver tissue [
155,
172]. In addition, higher amounts of ROS and ROS-mediated mtDNA damage have been described in patients with NASH compared to those without NASH [
51,
173]. Increased ROS levels have been reported to directly promote inflammation by activating inflammatory signaling pathways such as the NF-κB and JNK pathways as well as indirectly by increasing the gene expression of inflammatory cytokines such as TNF-α, TGF-β, and Fas ligand [
174]. TNF-α has been found to stimulate the release of cytochrome C from the mitochondria and leak into the cytosol, causing both apoptosis and necrosis [
175].
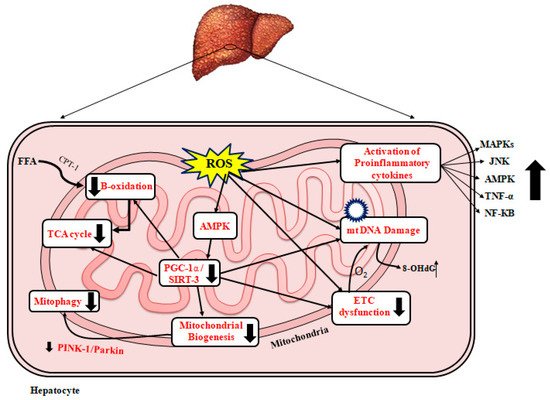
Figure 2 represents a schematic diagram of the effects of ROS leading to mitochondrial dysfunction and the development/progression of NAFLD, mitochondrial biogenesis, and NAFLD
Figure 2. Mitochondrial dysfunction and NAFLD. A schematic diagram representing the effect of oxidative stress leading to mitochondrial dysfunction in liver leading to NAFLD. FFA: free fatty acid; TCA: tricarboxylic acid cycle; ROS: reactive oxygen species; CPT-1: carnitine palmitoyl transferase 1; AMPK: AMP-activated protein kinase; PGC-1α: peroxisome proliferator-activated receptor-alpha; ETC: electron transport chain; SIRT3: sirtuin 3; MAPKs: mitogen-activated protein kinase; TNF-α: tumor necrosis factor-α; NF-KB: nuclear factor-KB, IL: Interleukin; JNK: c-jun N terminal kinase; 8-hydroxydeoxyguanosine; O2: oxygen; Pink: PTEN-induced kinase 1.