Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Mitochondrial dysfunction is a pathophysiological hallmark of most neurodegenerative diseases. Several clinical trials targeting mitochondrial dysfunction have been performed with conflicting results. Reliable biomarkers of mitochondrial dysfunction in vivo are thus needed to optimize future clinical trial designs. Neuroimaging methods could map in vivo changes of OXPHOS and oxidative stress, which are based on various physical phenomena to generate molecule-specific image contrasts.
- Neuroimaging
- Mitochondrial Dysfunction
- Metabolism
1. What Is Mitochondrial Impairment? The Molecular Complexity of a Fundamental Cell Organelle
Bioenergetic disturbances in the nervous system have been identified as a pathophysiological hallmark in many neurodegenerative diseases (NDs), including idiopathic Parkinson’s disease (PD) [1], atypical Parkinson’s disease (APD) [2], Alzheimer’s disease (AD) [3], among other forms of dementia, Huntington’s disease (HD) [4], prion disease [5], motor neuron disease (MND) [6], and certain forms of ataxia [7]. Most metabolic pathways (e.g., the tricarboxylic acid cycle, TCA) converge to the mitochondria and thus have an impact on the final steps of energy production by oxidative phosphorylation (OXPHOS) [1]. Yet, considering mitochondrial dysfunction purely by the resulting bioenergetic disturbances would be an oversimplification [1]. As a fundamental cellular organelle, mitochondria have widespread interconnections to the overall cellular homeostasis, making elucidating precise disease mechanisms of NDs challenging [8]. In addition, our current understanding of the molecular mechanisms underlying most NDs is incomplete and likely involves many pathophysiological events and processes [9]. Whether mitochondrial dysfunction can be considered the primary driver or simply a bystander of neurodegeneration largely remains to be elucidated [10]. Besides the overall complexity of disease mechanisms in NDs, mitochondrial dysfunction can refer to various unphysiological molecular processes [1]. These processes extend but are not limited to impaired mitochondrial biogenesis, dynamics and trafficking, calcium and metal ion dyshomeostasis, heme biosynthesis, control of cell division and cell fate decisions, and neuroinflammation [1]. The many-faceted dysfunction of mitochondria ultimately results in impaired energy supply and increased oxidative stress—both mechanisms leading to neurodegeneration, disease manifestation, and progression [8]. Therefore, these mechanisms could serve as viable surrogate markers for mitochondrial dyshomeostasis and can be mapped by innovative neuroimaging methods.
2. Why Could It Be Helpful to Identify Patients with Predominant Mitochondrial Dysfunction? On the Leap to Individualized Treatment Decisions
NDs are debilitating diseases, substantially impacting the well-being of patients and caregivers. The rising prevalence provides a significant socioeconomic burden to aging populations [11] and cannot be solely explained by the shifting age structure in modern societies [12]. Only symptomatic treatments are available to date, and no drug has been shown to reveal any clinically relevant disease-modifying properties [13]. NDs follow an individual time course often preceded by a prodromal phase [14]. Substantial neuronal cell loss has already occurred in prodromal phases, not yet having reached a symptom-causing threshold to facilitate a clinically established diagnosis. However, it would be highly desirable to identify patients in the prodromal phase to start neuroprotective treatments before disabling symptoms have occurred [14]. For some NDs, this prodromal phase can be outlined by clinical criteria (e.g., for PD) [15] or by pre-symptomatic genetic testing (e.g., for HD) [16]. Nevertheless, identifying study participants in a prodromal phase causes ethical dilemmas in conceptualizing clinical trials. Clinical concepts of the prodromal phase also imply that not all identified individuals will manifest their suspected disease [14]. Drug candidates must meet exceptionally high safety standards to be considered for clinical testing in pre-diseased individuals. Accompanying diagnostics could substantially enrich the frequency of disease-converters to ensure clinical trial success [17]. In NDs, the symptom severity and disease progression can be highly individual [18,19,20]. In addition, clinical trials in NDs evaluate disease-modifying properties of candidate drugs following long interventional periods [14]. Pathophysiology-orientated biomarkers and adaptive clinical trial designs will substantially improve the efficiency of neuroprotective trials by identifying surrogate markers for treatment response. These considerations could enrich study participants suitable for targeted therapies in innovative clinical trial designs. Patients suffering from NDs with suspected mitochondrial dysfunction are likely the most promising candidates for such innovative clinical trials. Human metabolism is a highly dynamic system. Therefore, immediate treatment responses following mitochondria-targeted therapies (e.g., by the pharmacological enhancement of OXPHOS) could be dynamically mapped within the scope of adaptive trials. Subsequently, reliable data can be generated to test if the paraclinical improvement of mitochondrial dysfunction can result in clinically relevant disease modification. Even though our current understanding of disease biology is constantly expanding, the molecular events leading to NDs are complex. They involve an interplay of various molecular events within an individual patient and disease course [21]. The molecular heterogeneity of NDs makes it unlikely that a single drug target can recapitulate all pathophysiological hallmarks of a given disease entity at any given time. A combination of potentially disease-modifying treatment strategies will likely be combined in a tailored and highly individualized fashion [21]. Developing reliable and pathophysiology-orientated biomarkers is a compulsory prerequisite for individualized treatment regimes. However, the role of mitochondrial dysfunction in the development and progression of NDs is undisputed [22]. Genetic insights and pathway-based analyses help elucidate mitochondrial dysfunction’s complexity in NDs. We refer the reader to a review article illustrating how these approaches help identify potential treatment strategies in patients with genetic and non-genetic Parkinson’s disease as a neurodegenerative model disease [23]. Although the evolvement and the individual time course of suspected mitochondrial dysfunction in NDs are only poorly understood, mitochondria-targeted therapies can still be considered a viable treatment strategy for many of these disorders [8]. The convergence of other disease mechanisms on mitochondrial homeostasis may help to reduce the complexity of drug development and testing [24]. The unknown temporal dynamics of mitochondrial dysfunction may result in a cause or consequence dilemma for our current understanding of disease biology [25]. The vital role of mitochondrial homeostasis for neuronal survival does not necessarily require causality in identifying mitochondria as viable treatment targets. For example, repeat expansions in the HTT gene cause HD mainly by protein misfolding and the toxic aggregation properties of the mutated HTT protein [26]. Even though mitochondrial dysfunction may not be the primary cause of neurodegeneration, it can be considered a direct consequence thereof [4]. Restoring mitochondrial homeostasis in HD could improve neuronal survival in HD. The clinical and molecular intricacy of NDs results in a pressing need for established biomarkers of mitochondrial dysfunction [27]. These biomarkers will shed light on the unclear clinical and molecular temporal dynamics of disease onset and progression and improve our current understanding of relevant disease mechanisms and the efficacy of clinical trials by dynamically monitoring responses to mitochondria-targeted treatments. Some promising methods are already available to establish potential biomarkers of mitochondrial dysfunction in vivo.
3. The Unbundling of Metabolic Pathways: Mitochondria at the Convergence of Human Metabolism
Mammalian metabolism is highly interwoven. Most nutrients are broken down into intermediary metabolites of the TCA. They can enter OXPHOS via complex I (by NADH) or complex II (by FADH2) of the electron transport chain (ETC) [8]. The metabolic conflux to the ETC offers possibilities to measure mitochondrial impairment indirectly. Upstream metabolites (e.g., of the TCA cycle) can subsequently accumulate to impaired OXPHOS (see Figure 1).
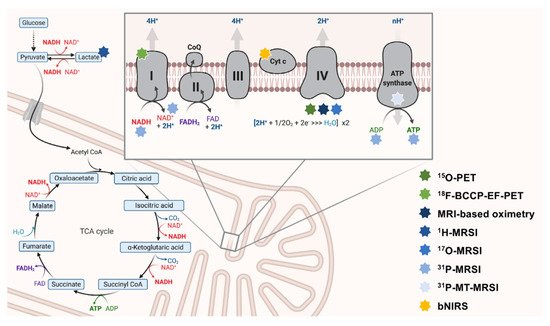
Figure 1. The interconnectedness of mitochondrial metabolism and OXPHOS-targeted neuroimaging approaches. Here, key aspects of OXPHOS are depicted. The ETC is schematically magnified (right upper corner). Multi-colored stars (legend: right-lower corner) indicate the respective neuroimaging modalities employed to map this particular aspect of metabolism. 18F-BCCP-EF: 2-tert-butyl-4-chloro-5-(6-(2-(2(18F)fluoroethoxy)-ethoxy]-pyridine-3-ylmethoxy)-2H-pyridazine-3-one. ADP: adenosine diphosphate. ATP: adenosine triphosphate. bNIRS: broadband near-infrared spectroscopy imaging. CoQ: coenzyme Q10. Cyt c: cytochrome c. ETC: electron transport chain. FAD/FADH2: flavin adenine dinucleotide. MRSI: magnetic resonance spectroscopy imaging. MT-MRSI: magnetization transfer magnetic resonance spectroscopy imaging. NAD+/NADH: nicotinamide adenine dinucleotide. OXPHOS: oxidative phosphorylation. PET: positron emission tomography. TCA: tricarboxylic acid.
Moreover, neurons may switch to non-OXPHOS catabolism to generate energy (e.g., by anaerobic glycolysis) [28]. In the past, in vivo measurements of lactate (as the end route of anaerobic glycolysis) have been evaluated to map mitochondrial impairment in patients with NDs [29] (see Figure 2).
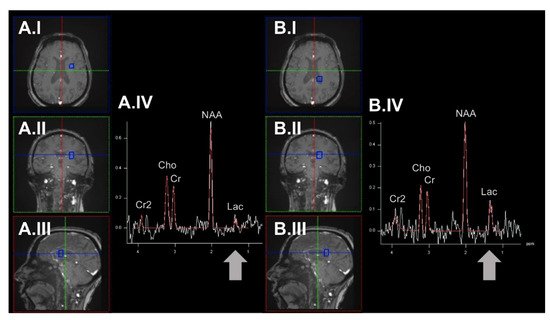
Figure 2. Exemplary determination of region-specific lactate levels by employing the 1H-MRSI methodology. Here, we demonstrate spectra derived from an example voxel (highlighted in blue) in the brain parenchyma of the left hemisphere (panel (A)) and the posterior portion of the left lateral ventricle (panel (B)). The respective voxel placement is shown in axial (A.I/B.I), coronal (A.II/B.II), and sagittal orientation (A.III/B.III). In this example, higher lactate levels can be observed in the CSF of the lateral ventricle. 1H-MRSI: proton magnetic resonance spectroscopy imaging. Cho: choline. Cr/Cr2: creatinine. CSF: cerebrospinal fluid. Lac: lactate. NAA: N-acetyl aspartate. ppm: parts per million.
Interestingly, other catabolic pathways (e.g., fatty acid breakdown by β-oxidation) are located in the mitochondria and can also be considered disease-relevant surrogate markers of mitochondrial dysfunction [30]. The interwovenness of mammalian metabolism also impacts the specificity of single molecules to quantify mitochondrial impairment in patients with NDs. In redox reactions, substrates of ETC complexes are often involved in various cellular pathways (e.g., NADH/ NAD+ as a ubiquitous coenzyme in redox reactions) [31]. The respective lack of pathophysiological specificity must be considered critically in conceptualizing studies probing mitochondrial impairment in NDs. However, these limitations extend to all studies on human metabolism and are not intrinsic to neuroimaging methods. Subsequently, quantifying single metabolites can only be interpreted as an estimation of in vivo mitochondrial impairment.
4. Neuroimaging for Patient Stratification and Therapy Monitoring in Patients with Suspected Mitochondrial Dysfunction?
The human brain accounts for only ~2% of the total body weight [32]. Based on the striking biomass difference between the human brain and the overall organism, it appears doubtful whether biomarkers derived from peripheral tissue (e.g., blood) can yield substantial diagnostic value in identifying NDs patients with cerebral mitochondrial dysfunction [1]. This is particularly important if only distinct neuronal subpopulations (e.g., hippocampal neurons in AD) are predominantly involved in disease development [33]. In marked contrast, the brain requires ~20% of the total cardiac output volume and ~25% of the human overall energy expenditure, pointing towards an exceptionally high metabolic demand [32]. Most of the organism’s metabolic activity is forwarded to the generation of ATP (by OXPHOS). Neuroimaging methods addressing the cerebral energy metabolism could therefore result in a high diagnostic yield to non-invasively identify patients with suspected mitochondrial dysfunction.
Interestingly, the study of brain energy metabolism has been one of the key interests of neuroscientists since the earliest neuroimaging methods have been available. For example, the biological origin of the blood-oxygen level-dependent imaging contrast (BOLD; frequently applied in functional MRI studies) has been elucidated for decades [34]. However, it took a substantial amount of time to advance this method to study in vivo oxygen consumption rates [35]. There seems to be a missing link between the exciting and rapidly expanding field of novel neuroimaging methods and the necessary translation into clinical practice. Indeed, there are significant challenges related to the clinical applicability of many of the following methods. We will discuss shortcomings and advancements required to facilitate the future applicability of these methods in clinical settings.
5. What Can We Measure? Mitochondrial Bioenergetics and Oxidative Stress as Promising Neuroimaging Markers of Mitochondrial Dysfunction
The synthesis of ATP by OXPHOS is recognized as the most prominent function of mitochondria. During OXPHOS, electrons can leak from the ETC and react with O2 to, e.g., form superoxides (O2−, one class of reactive oxygen species, ROS) [36]. Electron leakage constantly occurs during physiological conditions, and the resulting ROS are removed by various antioxidant-active coping mechanisms [36]. Mitochondrial dysfunction can negatively affect this process and severely disturb physiological OXPHOS [8]. The harmful elevation of ROS has several negative downstream effects, e.g., damaging proteins, which alter cellular homeostasis and impair neuronal survival [37]. The current understanding of NDs suggests that many pathophysiological hallmarks directly cause or result from OXPHOS disturbances and oxidative stress [8]. Therefore, the in vivo assessment of OXPHOS and oxidative stress appear viable targets for neuroimaging methods (see Figure 1). Besides OXPHOS and oxidative stress, other relevant pathophysiological aspects of NDs linked to mitochondrial dysfunction could be assessed by neuroimaging methods. Three prominent examples are:
Neuroimaging methods is to map in vivo changes of OXPHOS and oxidative stress. These methods are based on various physical phenomena to generate molecule-specific image contrasts. Here, three different physical phenomena were distinguished these methods based on:
-
Nuclear magnetic resonance (NMR). Atomic nuclei with a non-zero nuclear spin can be considered NMR-active. MRI and magnetic resonance spectroscopy (MRSI) utilize this phenomenon to generate image contrast or simultaneously measure multiple metabolites. In most MRI or MRSI studies, the NMR-phenomenon of protons (1H) is used to derive imaging data. However, these methods can also be applied to other NMR-active nuclei (e.g., 13C-, 15N-, 17O-, or 31P-nuclei). The use of non-1H nuclei in neuroimaging is often referred to as hetero-, multi-, or X-nuclear MR(S)I [38,45]. The acquisition of adequate X-nuclear MRSI signals critically depends on the natural abundance, the relative sensitivity (defined by the gyromagnetic ratio and the nuclear spin), and T1/T2 relaxation times (using quadrupolar relaxation) of the NMR-active isotope [38,46].
-
Near-infrared spectroscopy (NIRS). NIRS is a physical analysis technique based on the absorption and emission of short-waved (within the near-infrared region of the electromagnetic spectrum) light to detect chromophoric metabolites (e.g., hemoglobin) [47].
-
Radioactive decay. PET imaging is based on the simultaneous detection of two gamma-ray photons produced after a positron-emitting radionuclide (β+ decay) decay. The target-specific generation of radiotracers and the distribution of these weakly radioactively labeled substances can be used to image biochemical and physiological functions of the brain [48].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23137263
This entry is offline, you can click here to edit this entry!