Solid-state NMR is a structural biology approach that can provide information about large molecular assemblies at the atomic level. Contrary to solution-state NMR, it does not have any theoretical size limitation, and is performed on solids, i.e. sediments, crystals or powders which are filled into so-called 'NMR rotors'. To average some interactions, solid-state NMR requires to spin the rotors at the 'magic' angle and at high spinning frequencies. The NMR chemical shifts give a fingerprint of molecules, which can be used to characterize structural differences, and can be applied to different types of proteins such as viral capsids, membrane proteins, amyloid fibrils, etc...
- solid-state NMR
- protein NMR
- magic-angle spinning
- viral capsid
- NMR
- structural biology
- viral protein
1. Introduction
Solid-state NMR is a structural biology approach that can provide unique information about the molecular assemblies used by viruses to enter host cells, to establish infection and to ensure that progeny virions are released into the environment. Solid-state NMR can contribute to an understanding of how viral proteins and their assemblies function from a structural point of view, revealing weak spots which can be targeted to fight infection through antiviral therapies or vaccines. Solid-state NMR is in this respect complementary to solution NMR, X-ray crystallography and electron cryo-microscopy (cryo-EM), the other atomic-resolution structural biology methods. All can provide images of proteins at resolutions on the order of 1–10 Å. Considering large assemblies, X-ray crystallography was the first approach used to resolve viral capsid and envelope structures at high resolution, and was pioneered by Michael Rossmann and Stephen C. Harrison on plant viruses [1][2], owing to the enormous numbers of progeny viruses per cell that plants produce. Advances in recombinant DNA as well as cell culture technology opened the way to mammalian viruses [3][4]. This approach has subsequently revealed a wide variety of capsid structures, amongst them capsids from rhinovirus [5], poliovirus [6] and hepatitis B virus (HBV) [7], as other early examples of human pathogens. The typical resolution of the structures is around 2–3 Å, but a higher resolution below 1.5 Å has been reached for several viruses (see www.pdb.org). For X-ray structures, at 2.5 Å, the backbone is well-defined and many, though not all, sidechains are visible; at 1.5 Å, most sidechains are well-defined (and in the correct rotameric state) and some hydrogens including water are resolved [8][9]. While the resolution characterizing X-ray, cryo-EM and NMR structures cannot be directly compared, the numbers that they deliver still have a roughly similar meaning.
Resolving capsid structures by X-ray crystallography is, however, frequently impeded by the difficulties in obtaining highly diffracting crystals from these large, often flexible complexes, and by the requirement for milligram amounts of material for crystallization trials. Much of the available structural information has thus been gathered on the soluble subunits of the capsids, called the capsid or core proteins, which might in the crystal assemble into sub-particulate superstructures such as hexamers. This approximates how capsid proteins build hexagonal lattices, which are the foundation of icosahedral capsids [10][11]. Rossmann, besides using crystallography, was one of the first to recognize that a hybrid approach combining the moderate resolution structures from cryo-EM with high-resolution X-ray structures could allow to reconstitute the entire capsid assembly [12]. EM electron density maps, even at lower resolution, are also helpful since they can be used as an initial phasing model for the X-ray structure determination. Today, this follows standard protocols [13], and in addition to the first crystal structures, this highly successful combination approach has largely shaped our view on a large variety of viral capsids. EM thus had, from the beginning, an important share in structural virology, but was for a long time severely limited by the resolution attainable with the available technology. Still, even low resolution maps yielded in some cases structures; the HBV capsid is one of the first examples of a protein whose fold was determined from cryo-EM, although assisted by numerous biochemically derived constraints [14][15].
With the advent of direct detectors, cryo-EM underwent the “resolution revolution” [16], enabling similarly high resolution as X-ray and NMR which, again for the HBV capsid as example, reached 2.8 Å [17]. This step forward was central for structural virology, since it allowed for the first time to look at authentic viruses with atomic resolution, and to investigate structures of virus-like particles in different maturation states, as documented by numerous structures deposited in the PDB. The impressive pictures thus obtained for many viruses, including human pathogens such as Dengue virus (DENV), ZIKA virus, HBV and Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), many of them from Rossmann and coworkers, have shaped our view of infection. Moreover, the high resolution now allows to resolve the arrangement of proteins in challengingly large unit cells, as shown by the impressive 3.5 Å structure of a giant virus capsid, recently reported by Rossmann′s lab, which comprises numerous different proteins which intimately interact to form the capsid shell [18].
However, obtaining homogeneous samples from virus-replicating cells can be very demanding, owing to the often complex mixture of viral particles in different maturation states that coexist in such cells. Single particles can be picked on electron micrographs, allowing to exclude poorly assembled particles and/or to separate clearly different ones, such as the 180 subunit form (triangulation number T = 3) vs. the 240 subunit form (T = 4) of the HBV capsid. This approach has also been applied to capsids of duck HBV (DHBV), which in their larger core protein feature an extra domain that can adopt disordered and folded states [19]. Still, the differences must be large enough to enable separation of the different particle states, otherwise the resulting heterogeneity will manifest itself as a loss of resolution, and tracking different states remains difficult if not impossible.
This is where solid-state NMR comes into play, as it can contribute to aspects where X-ray crystallography and EM have limitations. Two strengths of solid-state NMR are of particular interest. First, conformational changes can be followed with highest sensitivity via the NMR chemical shifts (very small changes in protein conformation lead to considerable changes in resonance positions). Once the sequential assignment of a reference form is established, this allows to rapidly assess conformational variations as they occur, for instance, upon binding of an interaction partner to the reference structure. In fact, no full structure determination is needed to identify such conformational alterations, which makes the direct observation and quantification of structural transitions one of the strengths of NMR. The second major asset of NMR is its unrivaled ability to follow dynamic events over a wide range of experimental timescales that are too fast for cryo-EM and X-ray approaches, i.e., from picoseconds to seconds. NMR works under near-physiological conditions, and does not need the commonly very low temperatures of X-ray or cryo-EM, where mixed states of a sample are frozen out. Hence defining structural dynamics by these techniques often requires the availability of stable mimics of two or more different states.
2. Solid-State NMR
Solid-state NMR is a relative newcomer to the structural virology field, as sufficient spectral resolution and sensitivity to address complex proteins with atomic resolution have only recently been developed. Liquid-state (“in-solution”) and solid-state NMR in fact started out at the same time in the laboratories of Bloch and Purcell [20][21], and the spectra looked remarkably similar [20]. Historically, it was the development of sophisticated pulse techniques and multidimensional spectroscopy in the 1970s and 80s [22][23][24] which set the stage for protein structure determination by solution-state NMR [25]. Then, technical improvements in the homogeneity, stability and strengths of the magnetic fields applied dramatically increased resolution in the spectra of complex biomolecules for liquid-state NMR. Indeed, in solution the anisotropic interactions in the spin system are averaged by molecular tumbling and thus the “natural linewidth” is narrow (a few Hertz). This provides a high enough resolution in order to distinguish hundreds of resonances as they occur in medium-sized proteins. A natural limitation is set by the molecular tumbling which, today, restricts in solution NMR the maximum molecular mass for meaningful analyses of rigid proteins to roughly 50 kDa. Notably, this limit can be significantly pushed up by selective methyl-group protonation, which dilutes the dipolar coupling network; this allows, for example, investigation into the dynamics of proteins with a higher molecular weight, such as the 670 kDa proteasome [26].
In contrast, protein solid-state NMR was long plagued by much broader lines (around 50 kHz, and thus more than a thousand times broader than in solution), in particular for protons. This is due to the absence of molecular tumbling, which results in orientation-dependent anisotropic contributions to the chemical shifts, and the presence of a strong dipolar-coupling network (see Figure 1). One early conceptual break-through was the observation that mechanical rotation of the sample narrows the resonance lines. The strongest effect is found if the rotation axis is inclined from the magnetic field direction by the “magic” angle of θ ≈ 54.7° [27] for which the second-order Legendre polynomial, P2(cos θ) equals zero. By this, magic-angle spinning (MAS) removes the anisotropic contribution to the chemical shift and the dipole couplings if the rotation frequency is roughly of the same value as the size of the interaction to be averaged. The spectra then become liquid-like. The strengths of the interactions depends on their type, as well as the nuclei involved. While 13C chemical shift anisotropies are, for instance, on the order of a few kHz, and already very low spinning frequencies are sufficient to average them, it is highly beneficial to spin even faster than 100 kHz to average the strong proton–proton dipolar interactions. These approaches are currently under development [28][29].
Practical realization took a long time until improvements in 13C-detected spectroscopy led to the first high-resolution protein spectra that finally could be assigned [30] and be used for the first protein structure determination by solid-state NMR [31]. Early, proton detection had been applied at moderate spinning frequencies (40–60 kHz) to proteins using extensive deuteration to reduce proton–proton dipolar interactions [32][33]. Only in 2014, MAS frequencies finally exceeded 100 kHz in protein spectroscopy [34]. Indeed, this increase in MAS frequencies substantially increased resolution in deuterated proteins, and allowed for high-resolution 1H-detected spectroscopy also on fully protonated solid proteins, through more efficient averaging of the strong 1H-1H dipolar couplings. Detecting on 1H spins (as is routinely done in solution-state NMR) is more sensitive than 13C detection, mainly through the higher gyromagnetic ratio of the proton when compared to the 13C. This brought an almost 100-fold increase in mass sensitivity [35], reducing sample requirements to below 1 mg of protein and thus opening new possibilities in applications to biomolecules.
Still, the solid-state linewidth is typically somewhat broader than in solution-state NMR, which results in a higher spectral crowding than in solution, and limits the protein size to be investigated. Thus, further improvements in MAS frequency (>100 kHz [29][30][31][32][33][34][35][36]) and magnetic field strength (>800 MHz or 18.8 Tesla; note that magnetic field strength is conventionally expressed in units of the proton resonance frequency) will be important for the study of larger proteins. Typically, going from 800 MHz to 1.2 GHz increases the signal-to-noise ratio by nearly a factor of 2. But already today, the combination of 100 kHz MAS, high magnetic fields (>800 MHz) (Figure 1) and improved sample preparation, e.g., by direct sedimentation [37][38], allows for high-resolution spectroscopy of large biomolecular assemblies like viral capsids or envelopes [35][39][40][41] if they show high local symmetry. If the protein-monomers forming the assemblies are structurally diverse, this results in the observation of multiple sets of NMR signals (see also below), and increases spectral crowding or line widths if the splittings are unresolved.
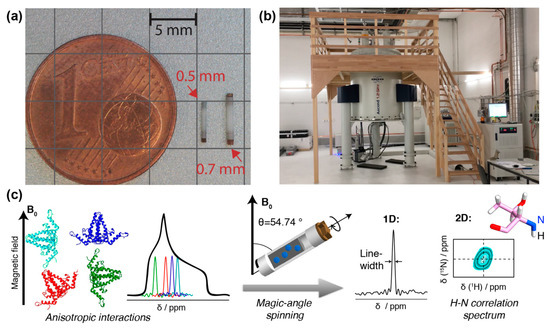
Figure 1. NMR rotors, spectrometer and resonance signals: (a) NMR rotors for fast spinning: 110 kHz for 0.7 mm rotor, 150 kHz for 0.5 mm (picture courtesy of Susanne Penzel). (b) 1200 MHz magnet for solid-state NMR at ETH Zurich. (c) Schematic representation of anisotropic interactions arising from different orientation of the individual molecules with respect to the magnetic field. The superposition of all possible chemical shifts gives rise to broad peaks with a characteristic shape, the powder pattern. The anisotropic interactions can be averaged out using MAS, which results in a single resonance line centered at the isotropic chemical shift of the spin. In addition, spinning sidebands may appear. This spin can be correlated to a neighboring spin, in the example, the amide 1H to the amide 15N. Two-dimensional spectroscopy then shows peaks which represent, for the present example, the amide proton frequency in one dimension, and the nitrogen frequency in the other. Such a signal will be observed for every NH pair in the protein. The resonance-line position is given by the isotropic part of NMR chemical shift and is usually specified in ppm (parts per million) of the resonance frequency. To obtain high-resolution spectra, the linewidth should be as narrow as possible, as this allows to distinguish (resolve) a maximum of resonances. A narrowing of linewidths can be achieved by (i) using a spectrometer operating at a higher magnetic field, (ii) improving sample homogeneity and symmetry, and (iii) reducing the dipolar coupling interactions by decreasing the density of protons in the system (through protein deuteration, for example) and/or increasing the MAS frequency by using smaller diameter rotors.
Solid-state NMR is thus a technique which can address structure and detailed conformation of proteins which are “not soluble”. This term must be understood in the broader sense, often simply meaning that they are involved in molecular interactions leading to larger superstructures, either with lipids for membrane proteins, or with each other, as in molecular assemblies such as molecular machines, protein fibrils, viral capsids or envelopes. “Solid” in this context means that the proteins do not show fast enough molecular tumbling (with correlation times of tens of nanoseconds) to be addressed by solution-state NMR. In a first approximation, therefore, the most attractive targets for solid-state NMR are large assemblies of many relatively small subunits, the latter being important to avoid excessive signal overlap in the spectra.
As mentioned, an additional important feature of solid-state NMR is its ability to characterize dynamic processes by observing relaxation times and exchange effects [42]. For an NMR spectroscopist, dynamics is usually associated with a timescale (the correlation time) and an amplitude (the order parameter), while structural virology dynamics is more associated with the volume of the conformational space for the protein. Dynamics on the pico- and nanosecond timescale (backbone and sidechain motion) can be characterized by relaxation measurements, while microsecond dynamics often leads to very efficient relaxation. In solid-state NMR spectra, dynamic entities (often loops but sometimes entire domains) may completely vanish from the spectrum as a consequence of such dynamics [43]. While limiting the possibilities for structure determination, the ability to assign a defined range of motional timescale to the corresponding residues provides valuable dynamic information.
As in solution-state NMR, the chemical shifts in MAS solid-state NMR give a fingerprint of a protein, which can be used to easily assess structural differences arising, for example, in the presence of interactants. This is of central importance in viral proteins, which often show multiple functions and thus also interactions, during which they change conformation to adapt to the specific role they need to fulfill at a specific point in the viral life cycle.
This entry is adapted from the peer-reviewed paper 10.3390/v12101069
References
- Hopper, P.; Harrison, S.C.; Sauer, R.T. Structure of tomato bushy stunt virus. V. Coat protein sequence determination and its structural implications. Mol. Biol. 1984, 177, 701–713.
- Silva, A.M.; Rossmann, M.G. Refined structure of southern bean mosaic virus at 2.9 A resolution. Mol. Biol. 1987, 197, 69–87.
- Krishnaswamy, S.; Rossmann, M.G. Structural refinement and analysis of Mengo virus. Mol. Biol. 1990, 211, 803–844.
- Arnold, E.; Rossmann, M.G. The use of molecular-replacement phases for the refinement of the human rhinovirus 14 structure. Acta Crystallogr. A, Found. Crystallogr. 1988, 44, 270–282.
- Kim, S.S.; Smith, T.J.; Chapman, M.S.; Rossmann, M.C.; Pevear, D.C.; Dutko, F.J.; Felock, P.J.; Diana, G.D.; McKinlay, M.A. Crystal structure of human rhinovirus serotype 1A (HRV1A). Mol. Biol. 1989, 210, 91–111.
- Filman, D.J.; Syed, R.; Chow, M.; Macadam, A.J.; Minor, P.D.; Hogle, J.M. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 1989, 8, 1567–1579.
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G. The crystal structure of the human hepatitis B virus capsid. Cell 1999, 3, 771–780.
- Elias, M.; Liebschner, D.; Koepke, J.; Lecomte, C.; Guillot, B.; Jelsch, C.; Chabriere, E. Hydrogen atoms in protein structures: High-resolution X-ray diffraction structure of the DFPase. BMC Res. Notes 2013, 6, 308–7.
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen atoms can be located accurately and precisely by x-ray crystallography. Adv. 2016, 2, e1600192, doi:10.1126/sciadv.1600192.
- Packianathan, C.; Katen, S.P.; Dann, C.E.; Zlotnick, A. Conformational Changes in the Hepatitis B Virus Core Protein Are Consistent with a Role for Allostery in Virus Assembly. Virol. 2010, 84, 1607–1615.
- Klumpp, K.; Lam, A.M.; Lukacs, C.; Vogel, R.; Ren, S.; Espiritu, C.; Baydo, R.; Atkins, K.; Abendroth, J.; Liao, G.; et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Natl. Acad. Sci. USA 2015, 112, 15196–15201.
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290.
- Liu, Z.; Guu, T.S.Y.; Cao, J.; Li, Y.; Cheng, L.; Tao, Y.J.; Zhang, J. Structure determination of a human virus by the combination of cryo-EM and X-ray crystallography. Biophys Rep. 2016, 2, 55–68.
- Böttcher, B.; Wynne, S.A.; Crowther, R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 1997, 386, 88–91.
- Conway, J.F.; Cheng, N.; Zlotnick, A.; Wingfield, P.T.; Stahl, S.J.; Steven, A.C. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 1997, 386, 91–94.
- Kühlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444.
- Böttcher, B.; Nassal, M. Structure of Mutant Hepatitis B Core Protein Capsids with Premature Secretion Phenotype. Mol. Biol. 2018, 430, 4941–4954.
- Fang, Q.; Zhu, D.; Agarkova, I.; Adhikari, J.; Klose, T.; Liu, Y.; Chen, Z.; Sun, Y.; Gross, M.L.; Van Etten, J.L.; et al. Near-atomic structure of a giant virus. Commun. 2019, 10, 388–11.
- Makbul, C.; Nassal, M.; Böttcher, B. Slowly folding surface extension in the prototypic avian hepatitis B virus capsid governs stability. eLife 2020, 9, doi:10.7554/eLife.57277.
- Bloch, F.; Hansen, W.W.; Packard, M. The Nuclear Induction Experiment. Rev. 1946, 70, 474–485.
- Purcell, E.M.; Torrey, H.C.; Pound, R.V. Resonance Absorption by Nuclear Magnetic Moments in a Solid. Rev. 1946, 69, 37–38.
- Aue, W.P.; Bartholdi, E.; Ernst, R.R. Two‐dimensional spectroscopy. Application to nuclear magnetic resonance. Chem. Phys. 1976, 64, 2229–2246.
- Jeener, J.; Meier, B.H.; Bachmann, P.; Ernst, R.R. Investigation of exchange processes by two‐dimensional NMR spectroscopy. Chem. Phys. 1979, 71, 4546–4553.
- Jeener, J.; Alewaeters, G. “Pulse pair technique in high resolution NMR” a reprint of the historical 1971 lecture notes on two-dimensional spectroscopy. Nucl. Mag. Res. Spectrosc. 2016, 94–95, 75–80.
- Wüthrich, K. NMR studies of structure and function of biological macromolecules (Nobel lecture). Angewandte Chemie Int. Ed. Engl. 2003, 42, 3340–3363.
- Religa, T.L.; Ruschak, A.M.; Rosenzweig, R.; Kay, L.E. Site-directed methyl group labeling as an NMR probe of structure and dynamics in supramolecular protein systems: Applications to the proteasome and to the ClpP protease. Am. Chem. Soc. 2011, 133, 9063–9068.
- Andrew, E.R.; Bradbury, A.; Eades, R.G. Removal of Dipolar Broadening of Nuclear Magnetic Resonance Spectra of Solids by Specimen Rotation. Nature 1959, 183, 1802–1803.
- Zhang, Z.; Oss, A.; Org, M.-L.; Samoson, A.; Li, M.; Tan, H.; Su, Y.; Yang, J. Selectively Enhanced 1H-1H Correlations in Proton-Detected Solid-State NMR under Ultrafast MAS Conditions. Phys. Chem. Lett. 2020, 11, 8077–8083.
- Schledorn, M.; Malär, A.A.; Torosyan, A.; Penzel, S.; Klose, D.; Oss, A.; Org, M.-L.; Wang, S.; Lecoq, L.; Cadalbert, R.; et al. Protein NMR Spectroscopy at 150 kHz Magic‐Angle Spinning Continues To Improve Resolution and Mass Sensitivity. Biol. 2020, 115, 11519, doi:10.1002/cbic.202000341.
- McDermott, A.E.; Polenova, T.; Böckmann, A.; Zilm, K.W.; Paulsen, E.K.; Martin, R.W.; Montelione, G.T. Partial NMR assignments for uniformly (13C, 15N)-enriched BPTI in the solid state. Biomol. NMR 2000, 16, 209–219.
- Castellani, F.; van Rossum, B.; Diehl, A.; Schubert, M.; Rehbein, K.; Oschkinat, H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 2002, 420, 98–102.
- Linser, R.; Bardiaux, B.; Andreas, L.B.; Hyberts, S.G.; Morris, V.K.; Pintacuda, G.; Sunde, M.; Kwan, A.H.; Wagner, G. Solid-State NMR Structure Determination from Diagonal-Compensated, Sparsely Nonuniform-Sampled 4D Proton–Proton Restraints. Am. Chem. Soc. 2014, 136, 11002–11010.
- Barbet-Massin, E.; Pell, A.J.; Retel, J.S.; Andreas, L.B.; Jaudzems, K.; Franks, W.T.; Nieuwkoop, A.J.; Hiller, M.; Higman, V.; Guerry, P.; et al. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. Am. Chem. Soc. 2014, 136, 12489–12497.
- Agarwal, V.; Penzel, S.; Szekely, K.; Cadalbert, R.; Testori, E.; Oss, A.; Past, J.; Samoson, A.; Ernst, M.; Böckmann, A.; et al. De novo 3D structure determination from sub-milligram protein samples by solid-state 100 kHz MAS NMR spectroscopy. Angewandte Chemie Int. Ed. 2014, 53, 12253–12256.
- Lecoq, L.; Schledorn, M.; Wang, S.; Smith-Penzel, S.; Malär, A.A.; Callon, M.; Nassal, M.; Meier, B.H.; Böckmann, A. 100 kHz MAS Proton-Detected NMR Spectroscopy of Hepatitis B Virus Capsids. Mol. Biosci. 2019, 6, doi:10.3389/fmolb.2019.00058.
- Penzel, S.; Oss, A.; Org, M.-L.; Samoson, A.; Böckmann, A.; Ernst, M.; Meier, B.H. Spinning faster: Protein NMR at MAS frequencies up to 126 kHz. Biomol. NMR 2019, 128, 12620–11.
- Gardiennet, C.; Schütz, A.K.; Hunkeler, A.; Kunert, B.; Terradot, L.; Böckmann, A.; Meier, B.H. A Sedimented Sample of a 59 kDa Dodecameric Helicase Yields High-Resolution Solid-State NMR Spectra. Angewandte Chemie Int. Ed. 2012, 51, 7855–7858.
- Bertini, I.; Luchinat, C.; Parigi, G.; Ravera, E.; Reif, B.; Turano, P. Solid-state NMR of proteins sedimented by ultracentrifugation. Natl. Acad. Sci. USA 2011, 108, 10396–10399.
- Andreas, L.B.; Jaudzems, K.; Stanek, J.; Lalli, D.; Bertarello, A.; Le Marchand, T.; Cala-De Paepe, D.; Kotelovica, S.; Akopjana, I.; Knott, B.; et al. Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Natl. Acad. Sci. USA 2016, 113, 9187–9192.
- Wang, S.; Fogeron, M.-L.; Schledorn, M.; Dujardin, M.; Penzel, S.; Burdette, D.; Berke, J.M.; Nassal, M.; Lecoq, L.; Meier, B.H.; Böckmann; et al. A. Combining Cell-Free Protein Synthesis and NMR Into a Tool to Study Capsid Assembly Modulation. Mol. Biosci. 2019, 6, doi:10.3389/fmolb.2019.00067.
- David, G.; Fogeron, M.-L.; Schledorn, M.; Montserret, R.; Haselmann, U.; Penzel, S.; Badillo, A.; Lecoq, L.; André, P.; Nassal, M.; et al. A. Structural Studies of Self-Assembled Subviral Particles: Combining Cell-Free Expression with 110 kHz MAS NMR Spectroscopy. Angewandte Chemie Int. Ed. 2018, 57, 4787–4791.
- Schanda, P.; Ernst, M. Studying dynamics by magic-angle spinning solid-state NMR spectroscopy: Principles and applications to biomolecules. Nucl. Mag. Res. Sp. 2016, 96, 1–46.
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid Fibrils of the HET-s(218-289) Prion Form a Solenoid with a Triangular Hydrophobic Core. Science 2008, 319, 1523–1526.