Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Alzheimer’s disease (AD) is a progressive degenerative disorder and a leading cause of dementia in the elderly. The etiology of AD is multifactorial, including an increased oxidative state, deposition of amyloid plaques, and neurofibrillary tangles of the tau protein.
- Alzheimer’s Disease
- metabolism
- liver
- Exercise
1. Alzheimer’s Disease and Exercise
Great efforts have been made to find reliable prevention methods and develop drugs or disease-modifying therapies to treat Alzheimer’s disease (AD), yet without success [1][2][3][4]. These findings emphasize the importance of prevention. It is known that genes can predispose to AD [5][6], but it is also known that lifestyle habits are associated with the incidence of AD [7][8]. It has been reported that physical inactivity increases the incidence of AD [4]. Therefore, it is not surprising that physical activity can reduce the occurrence of AD [8]. Regular exercise has been shown to induce adaptation in the whole body, including the neuronal, cardiovascular, skeletal, immune, digestive, and reproductive systems [9][10]. Furthermore, training in AD (TAD) is known to have a positive impact on alteration of neurotrophin synthesis, attenuation of oxidative stress, inflammation, induction of amyloid-β (Aβ) degrading enzymes, an increase in vascularization and blood flow, and energy metabolism of the brain [8]. It is also known that exercise can reduce the concentration of Aβ in plasma [11] and can be protective against Aβ neurotoxicity with the disease [12].
2. Exercise and Liver with Alzheimer’s Disease
Increasing evidence suggests an important role of liver function in the pathophysiology of AD. Proteins, such as amyloid-β and hyperphosphorylated tau, are vital contributors to the onset or progression of AD. The liver is theoretically involved in the peripheral clearance of circulating Aβ in the blood [13].
In addition to the beneficial systemic effects, exercise positively impacts liver function in AD. For example, abnormal levels of liver enzymes associated with the diagnosis of Alzheimer’s and correlated with poor memory and thinking scores have been observed [14]. Furthermore, microbial changes caused by exercise and probiotic treatment alter liver metabolism, mitochondrial content, and antioxidant capacity in APP/PS1 transgenic mice. Training and probiotic supplementation did not significantly raise mitochondrial counts in the AD animal’s liver, based on cytochrome c oxidase subunit 4 (COX4) and peroxisome-proliferator-activated receptor–gamma coactivator 1 alpha (PGC-1α) protein levels. However, on the other hand, the mitochondrial antioxidant capacity changed positively due to regular exercise. Antioxidant signaling proteins, such as nuclear factor erythroid 2-related factor 2 (NRF-2) and superoxide dismutase 2 (SOD2), were also investigated. NRF-2, the major regulator of antioxidant protection, showed an increase in the group of trained and probiotic supplemented animals compared with the AD group. Furthermore, decreased superoxide SOD2 levels were observed in AD, while training prevented this alteration [15].
3. Exercise and Gonads with Alzheimer’s Disease
Testes are also peripheral organs affected by AD. In AD organs, decreased numbers of spermatogonia, spermatocytes, and interstitial Leydig cells have been observed. Immunoreactivity of collagen type IV in seminiferous tubules’ basement membrane (bm) was hardly detectable, and decreased bm thickness in AD testes, resulting in changes in blood–testes barrier function.
Studies show that testicular degradation can be compensated for by regular physical activity in a mouse model of AD [16]. In TAD animals, the number of convoluted seminiferous tubules’ cells was partially recovered, and the number of Leydig cells was elevated after physical activity. As a result of training, the thickness of the basement membrane became almost as thick as in WT mice. Expression of the type IV collagen molecule was also elevated, maintaining the integrity of the basal membrane, and thus compensating for the adverse effects of AD [17].
The PACAP, as mentioned above, is expressed in not only the CNS but also peripheral organs, with the highest level in testes [18]. PACAP regulation seems essential for maintaining the typical structure of testes and spermatogenesis and male sex hormone production [19]. It is proven that a lack of PACAP protein or a mistake in PACAP downstream signaling causes morphological and functional changes in testis [20]. The messenger RNA (mRNA) expression and the protein level of the PAC1R were decreased in AD animals compared with WT [17]. Interestingly, this reduction in PAC1R has also been demonstrated in the CNS of AD models [21]. In contrast, the mRNA and protein expression of VPAC1R and VPAC2R did not show a significant difference in AD. In the TAD experimental group, an increase in PAC1R and VPAC1R was noted, whereas a decrease in VPAC2 receptor protein expression was observed. The downstream PACAP signaling pathway elements, such as the cAMP, PKA, P-PKA, and PP2A protein expressions, were also significantly decreased in the samples of AD mice. In TAD testes, expression of the PKA protein was elevated almost to WT levels, and P-PKA and PPA2 were augmented considerably by physical activity [17].
There are few data available about ovary in AD. The most common metabolic disorder in premenopausal women is polycystic ovary syndrome (PCOS) [22]. Patients with PCOS have increased LH-FSH levels, decreased vitamin D and insulin resistance, and obesity [23]. These are important factors also in AD and may increase the risk of the disease [24]. Moderate exercise (guidelines for PCOS suggest at least 150 min of physical activity per week) is helpful in PCOS, so it can contribute to minimize the chance of developing AD [25]. Furthermore, women with premature menopause have an increased risk of AD [26]. Menopausal hormone therapy supplemented with regular training may give the chance to reduce the risk of AD in later life [27][28].
4. Exercise and Kidney in Alzheimer’s Disease
Pathological Aβ accumulation has also been observed in kidneys of AD animals leading to possible fibrosis, causing filtration disorders and renal insufficiency [29]. In AD mice, homogenous eosinophilic deposits in the tubular systems and strong Aβ positivity were visible in the kidneys [30].
Physical exercise has been shown to positively affect the morphology and function of kidneys in the AD mouse model. Training can reduce kidney fibrosis, which induces Aβ clearance and may help inhibit the disease’s progression [31]. First of all, exercise reduced Aβ accumulation and diminished eosinophilic deposits [30]. After physical activity in AD mice, the amount of interstitial collagen type I was reduced compared with the untrained group. Furthermore, both normalized mRNA and protein expression of collagen type I were measured in TAD animals [32]. Additionally, as a result of exercise, the immunopositivity and (mRNA and protein) the expression of collagen type IV were normalized/elevated in trained animals [30]. The normalized basement membrane formation can play a role in Aβ elimination via kidneys and help inhibit the disease’s progression [30][31]. Accordingly, collagen type IV has been observed to inhibit Aβ plaque formation [33].
Pathological fibrosis in the kidneys of AD animals raises the role of transforming the growth factor β (TGFβ) pathway [34][35]. TGFβ is an essential factor in the pathogenesis of AD in the brain and a master regulator of renal inflammation and fibrosis, consequently responsible for appropriate filtration [36][37]. TGFβ causes increased collagen expression and accumulation [38]. Physical activity, through TGFβ signalization, may prevent renal fibrosis and support Aβ clearance in the periphery. Activation of canonical and non-canonical TGFβ pathways was observed in AD, which was normalized in TAD mice. Although TGFβ1′s mRNA and protein expression did not significantly differ between WT and AD kidneys, increased expressions were found in TAD samples. As for the receptors, the expression of TGFβRI protein was reduced in AD mice and normalized after physical activity, while the expression of mRNA and protein of TGFβRII changed to the contrary [32].
TGFβ signaling interacts with the group of mitogen-activated protein kinases (MAPKs), such as extracellular signal-regulated kinase 1/2 (ERK1/2), p38 mitogen-activated protein kinase (p38), and Jun N-terminal kinase (JNK) [39]. Members of the MAPK family strongly support distinct functions in the pathogenesis of renal fibrosis. For example, ERK contributes to renal fibrotic transformation, whereas inhibition of ERK activity reduces interstitial fibrosis in AD [40]. The MAPK family shows alteration in the kidney of AD after long-term training. After physical activity, standard ERK and a more active form of the kinase, phospho-ERK expression, and normalized renal function were detected in TAD samples [32]. The protein expression of ERK1/2 kinase was reduced in the kidneys of AD mice, with normalization after physical exercise. The amount of the phosphorylated ERK was significantly elevated in AD samples and showed a significant decrease in TAD mice. The other member of the MAPK family is the p38 protein. Its expression was increased in AD mice, inducing fibrosis, while the phosphorylated form of p38 changed to the contrary. Exercise normalized p38 levels suggest a balancing function of training in p38-mediated fibrosis formation [32][41]. Therefore, inhibition of p38 may be a good target in fibrosis treatment [42]. Furthermore, JNK kinase also affects glomerular filtration [43], and the inhibition of the kinase may suppress interstitial fibrosis [44]. The protein expression of the two isoforms of JNK revealed a significant reduction in AD mice but showed elevation in TAD animals [32].
Matrix metalloproteinase 9 (MMP9) is also involved in fibrotic processes by degrading extracellular matrix elements such as type I, IV, and type V of collagen and other extracellular matrix proteins [45][46]. MMP9 is regulated by TGF signaling in the kidney, and its activation is p38-dependent [45][47]. Surprisingly, a significant increase in MMP9 protein expression was observed in the kidneys of AD mice, but MMP9 expression was dramatically increased after exercise, congruent with the marked decrease in collagen type I in the tubular system [32].
The TGF signaling pathway’s activation can also play an important role in AD pathogenesis through cell cycle/cellular proliferation and apoptosis [48]. Cell proliferation markers, such as cyclin-dependent kinase inhibitor 1 (CDKN1/p21) and proliferating cell nuclear antigen (PCNA) expressions, were measured in AD: CDKN1/p21 activation and substantial PCNA reductions in the tubular system of AD mice were normalized by physical activity [30]. The CDKN1/p21 elevation suggests a cell cycle arrest in AD, and CDKN1/p21 interaction with PCNA can block cells in the S-phase of the cell cycle [49]. Caspase activation can mediate via Aβ accumulation: the more active, cleaved form of caspase 3 appeared in AD samples, indicating increased apoptosis and the loss of tubular cell function, while physical activity reduced its expression [30][50].
The abovementioned neuroprotective PACAP also has a nephroprotective role in various renal pathologies [51]. PACAP is produced in kidneys, where the most dramatic amyloid deposition was found in PACAP knockout mice. This signal molecule may be one of the most promising targets for the renal elimination of Aβ [20]. Examining molecular processes in AD mice, the protein expressions of PAC1R, VPAC1R, and VPAC2R were demonstrated in WT kidneys. In contrast, the expressions of all PACAP receptors were almost undetectable or significantly decreased in AD samples. Interestingly, the level of all PACAP receptors increased in TAD mice: most dominantly, expressions of PAC1R and VPAC1R were elevated [30]. PACAP equally binds both PAC1R and VPAC1R in kidney diseases [52]. PACAP signaling pathway can be regulated through PKA [18]. In AD animals, reduced PKA protein expression was observed, almost normalized after physical activity [30]. The activation/phosphorylation of PKA increases the level of transcription factor cAMP-response-element-binding protein (CREB) [53], the protein level of which was also decreased (or its activated form was undetectable) in AD mice and augmented after exercise [30]. These data suggest PACAP and PKA signaling may be involved in “physical-activity-mediated defense mechanisms” in AD. Furthermore, PACAP also plays an essential role in bone morphogenetic protein (BMP) signaling [54], as does a member of the TGFβ superfamily [55]. Initially, BMP was identified as “only” an osteogenic factor, but nowadays, it is known that BMPs perform several other functions [55][56][57][58]. PACAP and BMP signaling is involved in preventing diseases through physical activity. For example, modified BMP/Smad signaling has been reported in AD mice [30][52]. BMPs and Smad transcription factors regulate the expression of genes associated with fibrosis and basement membrane components, such as collagen [59][60]. Both BMP receptor type 1 (BMPR1) mRNA and protein levels and the expression of bone morphogenetic protein 4 (BMP4), which induces Smad1, showed a noticeable reduction in AD samples. Still, they were significantly augmented as a result of training. Smad2 and Smad3 expressions were also altered in the kidneys of AD mice: Smad2 increased, while Smad3 showed a moderate decrease in AD samples, but physical activity can compensate for these alterations [30].
TGFβRII, ERK, p38, JNK, and Smad phosphorylation occur on serine (Ser) amino acid, which raises a possible activation of serine–threonine protein phosphatases (Ser/Thr phosphatases) in AD [61]. Altered PP2A and PP2B expressions and modified protein phosphorylations have been reported in AD [62][63][64]. PP2A can regulate tau phosphorylation with a higher affinity than PP2B [65]. Reduced PP2A, but an increased PP2B expression in kidneys of AD mice was observed, similarly to the CNS, which can be compensated by physical exercise [30]. Increased phosphorylation on Ser residues, as the result of PP2A expression decrease, can induce the hyperphosphorylation of tau protein or APP, as was predicted in AD. In addition, ERK kinase dephosphorylation can happen dominantly through PP2A activation [66]. Subsequently, reduced expression of PP2A can lead to increased ERK phosphorylation in AD [30]. The PP2B can directly regulate the dephosphorylation of JNK and p38 kinases [67]. Therefore, the decreased activation of these kinases is the consequence of elevated PP2B expression in AD (Figure 1).
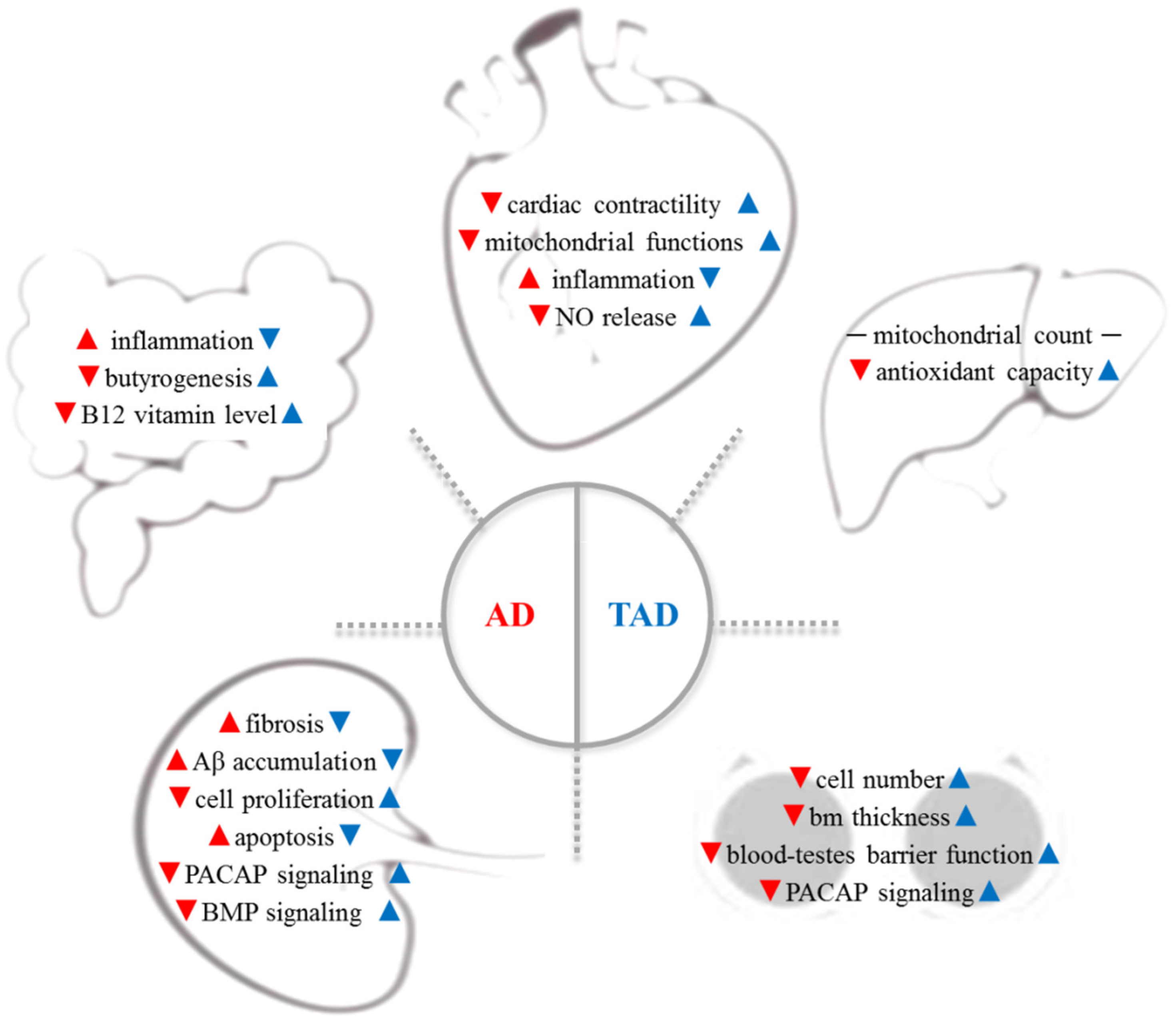
Figure 1. AD is a complex systemic disorder, which induces the degenerative process in organs distant from the brain: cardiovascular system, gut microbiome, liver, testes, and kidney are involved (red arrows). Therefore, increased physical activity has been reported to have a preventive effect on all organs in AD (blue arrows). Aβ—β-amyloid; AD—Alzheimer’s disease; bm—basement membrane; BMP—bone morphogenetic protein; NO—nitric oxide, PACAP—pituitary--cyclase-activating polypeptide; TAD—trained AD.
This entry is adapted from the peer-reviewed paper 10.3390/antiox11051028
References
- Panda, S.S.; Jhanji, N. Natural products as potential anti-Alzheimer agents. Curr. Med. Chem. 2020, 27, 5887–5917.
- Whitehouse, P.J. Ethical issues in early diagnosis and prevention of Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 101–108.
- Raggi, A.; Tasca, D.; Ferri, R. A brief essay on non-pharmacological treatment of Alzheimer’s disease. Rev. Neurosci. 2017, 28, 587–597.
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828.
- Tanzi, R.E.; George-Hyslop, P.S.; Gusella, J.F. Molecular genetics of Alzheimer disease amyloid. J. Biol. Chem. 1991, 266, 20579–20582.
- Price, D.L.; Borchelt, D.R.; Sisodia, S.S. Alzheimer disease and the prion disorders amyloid beta-protein and prion protein amyloidoses. Proc. Natl. Acad. Sci. USA 1993, 90, 6381–6384.
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639.
- Radak, Z.; Hart, N.; Sarga, L.; Koltai, E.; Atalay, M.; Ohno, H.; Boldogh, I. Exercise plays a preventive role against Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 777–783.
- Radak, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 2008, 44, 153–159.
- Radak, Z.; Torma, F.; Berkes, I.; Goto, S.; Mimura, T.; Posa, A.; Balogh, L.; Boldogh, I.; Suzuki, K.; Higuchi, M.; et al. Exercise effects on physiological function during aging. Free Radic. Biol. Med. 2019, 132, 33–41.
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 62, 101108.
- Meng, Q.; Lin, M.S.; Tzeng, I.S. Relationship between exercise and Alzheimer’s disease: A narrative literature review. Front. Neurosci. 2020, 14, 131.
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver dysfunction as a novel player in Alzheimer’s progression: Looking outside the brain. Front. Aging Neurosci. 2019, 11, 174.
- Nho, K.; Kueider-Paisley, A.; Ahmad, S.; MahmoudianDehkordi, S.; Arnold, M.; Risacher, S.L.; Louie, G.; Blach, C.; Baillie, R.; Han, X.; et al. Association of altered liver enzymes with Alzheimer disease diagnosis, cognition, neuroimaging measures, and cerebrospinal fluid biomarkers. JAMA Netw. Open 2019, 2, e197978.
- Téglás, T.; Ábrahám, D.; Jókai, M.; Kondo, S.; Mohammadi, R.; Fehér, J.; Szabó, D.; Wilhelm, M.; Radák, Z. Exercise combined with a probiotics treatment alters the microbiome, but moderately affects signalling pathways in the liver of male APP/PS1 transgenic mice. Biogerontology 2020, 21, 807–815.
- Torma, F.; Koltai, E.; Nagy, E.; Ziaaldini, M.M.; Posa, A.; Koch, L.G.; Britton, S.L.; Boldogh, I.; Radak, Z. Exercise increases markers of spermatogenesis in rats selectively bred for low running capacity. PLoS ONE 2014, 9, e114075.
- Szegeczki, V.; Horváth, G.; Perényi, H.; Tamás, A.; Radák, Z.; Ábrahám, D.; Zákány, R.; Reglodi, D.; Juhász, T. Alzheimer’s disease mouse as a model of testis degeneration. Int. J. Mol. Sci. 2020, 21, 5726.
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357.
- Prisco, M.; Rosati, L.; Morgillo, E.; Mollica, M.P.; Agnese, M.; Andreuccetti, P.; Valiante, S. Pituitary adenylate cyclase-activating peptide (PACAP) and its receptors in Mus musculus testis. Gen. Comp. Endocrinol. 2020, 286, 113297.
- Reglodi, D.; Jungling, A.; Longuespée, R.; Kriegsmann, J.; Casadonte, R.; Kriegsmann, M.; Juhasz, T.; Bardosi, S.; Tamas, A.; Fulop, B.D.; et al. Accelerated pre-senile systemic amyloidosis in PACAP knockout mice—A protective role of PACAP in age-related degenerative processes. J. Pathol. 2018, 245, 478–490.
- Han, P.; Tang, Z.; Yin, J.; Maalouf, M.; Beach, T.G.; Reiman, E.M.; Shi, J. Pituitary adenylate cyclase-activating polypeptide protects against β-amyloid toxicity. Neurobiol. Aging 2014, 35, 2064–2071.
- Spinedi, E.; Cardinali, D.P. The polycystic ovary syndrome and the metabolic syndrome: A possible chronobiotic-cytoprotective adjuvant therapy. Int. J. Endocrinol. 2018, 2018, 1349868.
- González, F. Inflammation in polycystic ovary syndrome: Underpinning of insulin resistance and ovarian dysfunction. Steroids 2012, 77, 300–305.
- Sarahian, N.; Sarvazad, H.; Sajadi, E.; Rahnejat, N.; Eskandari Roozbahani, N. Investigation of common risk factors between polycystic ovary syndrome and Alzheimer’s disease: A narrative review. Reprod. Health 2021, 18, 156.
- Woodward, A.; Klonizakis, M.; Broom, D. Exercise and polycystic ovary syndrome. Adv. Exp. Med. Biol. 2020, 1228, 123–136.
- Davey, D.A. Alzheimer’s disease, dementia, mild cognitive impairment and the menopause: A ‘window of opportunity’? Women’s Health 2013, 9, 279–290.
- Thomas, A.; Daley, A.J. Women’s views about physical activity as a treatment for vasomotor menopausal symptoms: A qualitative study. BMC Women’s Health 2020, 20, 203.
- Davey, D.A. Prevention of Alzheimer’s disease, cerebrovascular disease and dementia in women: The case for menopause hormone therapy. Neurodegener. Dis. Manag. 2017, 7, 85–94.
- Kheirbakhsh, R.; Haddadi, M.; Muhammadnejad, A.; Abdollahi, A.; Shahi, F.; Amanpour-Gharaei, B.; Abrahim-Habibi, A.; Barati, T.; Amanpour, S. Long-term behavioral, histological, biochemical and hematological evaluations of amyloid beta-induced Alzheimer’s disease in rat. Acta Neurobiol. Exp. 2018, 78, 51–59.
- Perényi, H.; Szegeczki, V.; Horváth, G.; Hinnah, B.; Tamás, A.; Radák, Z.; Ábrahám, D.; Zákány, R.; Reglodi, D.; Juhász, T. Physical activity protects the pathological alterations of Alzheimer’s disease kidneys via the activation of PACAP and BMP signaling pathways. Front. Cell. Neurosci. 2020, 14, 243.
- Ghiso, J.; Calero, M.; Matsubara, E.; Governale, S.; Chuba, J.; Beavis, R.; Wisniewski, T.; Frangione, B. Alzheimer’s soluble amyloid beta is a normal component of human urine. FEBS Lett. 1997, 408, 105–108.
- Szegeczki, V.; Perényi, H.; Horváth, G.; Hinnah, B.; Tamás, A.; Radák, Z.; Ábrahám, D.; Zákány, R.; Reglodi, D.; Juhász, T. Physical training inhibits the fibrosis formation in Alzheimer’s disease kidney influencing the TGFβ signaling pathways. J. Alzheimer’s Dis. 2021, 81, 1195–1209.
- Kiuchi, Y.; Isobe, Y.; Fukushima, K. Type IV collagen prevents amyloid beta-protein fibril formation. Life Sci. 2002, 70, 1555–1564.
- Kajdaniuk, D.; Marek, B.; Borgiel-Marek, H.; Kos-Kudła, B. Transforming growth factor β1 (TGFβ1) in physiology and pathology. Endokrynol. Pol. 2013, 64, 384–396.
- Chen, L.; Yang, T.; Lu, D.W.; Zhao, H.; Feng, Y.L.; Chen, H.; Chen, D.Q.; Vaziri, N.D.; Zhao, Y.Y. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 2018, 101, 670–681.
- Grammas, P.; Ovase, R. Cerebrovascular transforming growth factor-beta contributes to inflammation in the Alzheimer’s disease brain. Am. J. Pathol. 2002, 160, 1583–1587.
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491.
- Browne, J.A.; Liu, X.; Schnaper, H.W.; Hayashida, T. Serine-204 in the linker region of Smad3 mediates the collagen-I response to TGF-β in a cell phenotype-specific manner. Exp. Cell Res. 2013, 319, 2928–2937.
- Ma, F.Y.; Sachchithananthan, M.; Flanc, R.S.; Nikolic-Paterson, D.J. Mitogen activated protein kinases in renal fibrosis. Front. Biosci. 2009, 1, 171–187.
- Wu, Y.; Wang, L.; Deng, D.; Zhang, Q.; Liu, W. Renalase protects against renal fibrosis by inhibiting the activation of the ERK signaling pathways. Int. J. Mol. Sci. 2017, 18, 855.
- Lee, J.; An, J.N.; Hwang, J.H.; Lee, H.; Lee, J.P.; Kim, S.G. p38 MAPK activity is associated with the histological degree of interstitial fibrosis in IgA nephropathy patients. PLoS ONE 2019, 14, e0213981.
- Stambe, C.; Atkins, R.C.; Tesch, G.H.; Masaki, T.; Schreiner, G.F.; Nikolic-Paterson, D.J. The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J. Am. Soc. Nephrol. 2004, 15, 370–379.
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling pathway in renal fibrosis. Front. Physiol. 2017, 8, 829.
- Ma, F.Y.; Flanc, R.S.; Tesch, G.H.; Han, Y.; Atkins, R.C.; Bennett, B.L.; Friedman, G.C.; Fan, J.H.; Nikolic-Paterson, D.J. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J. Am. Soc. Nephrol. 2007, 18, 472–484.
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306.
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Models Mech. 2014, 7, 193–203.
- Zhao, H.; Dong, Y.; Tian, X.; Tan, T.K.; Liu, Z.; Zhao, Y.; Zhang, Y.; Harris, D.; Zheng, G. Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases. World J. Nephrol. 2013, 2, 84–89.
- Liu, Y.; Shang, D. Transforming growth factor-β1 enhances proliferative and metastatic potential by up-regulating lymphoid enhancer-binding factor 1/integrin αMβ2 in human renal cell carcinoma. Mol. Cell. Biochem. 2020, 465, 165–174.
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306.
- Park, G.; Nhan, H.S.; Tyan, S.H.; Kawakatsu, Y.; Zhang, C.; Navarro, M.; Koo, E.H. Caspase activation and caspase-mediated cleavage of APP is associated with amyloid β-protein-induced synapse loss in Alzheimer’s disease. Cell Rep. 2020, 31, 107839.
- Horvath, G.; Opper, B.; Reglodi, D. The neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) is protective in inflammation and oxidative stress-induced damage in the kidney. Int. J. Mol. Sci. 2019, 20, 4944.
- Li, M.; Maderdrut, J.L.; Lertora, J.J.; Arimura, A.; Batuman, V. Renoprotection by pituitary adenylate cyclase-activating polypeptide in multiple myeloma and other kidney diseases. Regul. Pept. 2008, 145, 24–32.
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609.
- Pavelock, K.A.; Girard, B.M.; Schutz, K.C.; Braas, K.M.; May, V. Bone morphogenetic protein down-regulation of neuronal pituitary adenylate cyclase-activating polypeptide and reciprocal effects on vasoactive intestinal peptide expression. J. Neurochem. 2007, 100, 603–616.
- Bandyopadhyay, A.; Yadav, P.S.; Prashar, P. BMP signaling in development and diseases: A pharmacological perspective. Biochem. Pharmacol. 2013, 85, 857–864.
- Józsa, G.; Fülöp, B.D.; Kovács, L.; Czibere, B.; Szegeczki, V.; Kiss, T.; Hajdú, T.; Tamás, A.; Helyes, Z.; Zákány, R.; et al. Lack of pituitary adenylate cyclase-activating polypeptide (PACAP) disturbs callus formation. J. Mol. Neurosci. 2021, 71, 1543–1555.
- Laszlo, E.; Juhasz, T.; Varga, A.; Czibere, B.; Kovacs, K.; Degrell, P.; Horvath, G.; Jancso, G.; Szakaly, P.; Tamas, A.; et al. Protective effect of PACAP on ischemia/reperfusion-induced kidney injury of male and female rats: Gender differences. J. Mol. Neurosci. 2019, 68, 408–419.
- Józsa, G.; Szegeczki, V.; Pálfi, A.; Kiss, T.; Helyes, Z.; Fülöp, B.; Cserháti, C.; Daróczi, L.; Tamás, A.; Zákány, R.; et al. Signalling alterations in bones of pituitary adenylate cyclase activating polypeptide (PACAP) gene deficient mice. Int. J. Mol. Sci. 2018, 19, 2538.
- Von Bubnoff, A.; Cho, K.W. Intracellular BMP signaling regulation in vertebrates: Pathway or network? Dev. Biol. 2001, 239, 1–14.
- Matsubara, T.; Araki, M.; Abe, H.; Ueda, O.; Jishage, K.; Mima, A.; Goto, C.; Tominaga, T.; Kinosaki, M.; Kishi, S.; et al. Bone morphogenetic protein 4 and Smad1 mediate extracellular matrix production in the development of diabetic nephropathy. Diabetes 2015, 64, 2978–2990.
- Heldin, C.H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053.
- Qian, W.; Yin, X.; Hu, W.; Shi, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X.; Liu, F. Activation of protein phosphatase 2B and hyperphosphorylation of tau in Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 23, 617–627.
- Oliveira, J.M.; Henriques, A.G.; Martins, F.; Rebelo, S.; da Cruz e Silva, O.A. Amyloid-β modulates both AβPP and tau phosphorylation. J. Alzheimer’s Dis. 2015, 45, 495–507.
- Leong, W.; Xu, W.; Wang, B.; Gao, S.; Zhai, X.; Wang, C.; Gilson, E.; Ye, J.; Lu, Y. PP2A subunit PPP2R2C is downregulated in the brains of Alzheimer’s transgenic mice. Aging 2020, 12, 6880–6890.
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927.
- Adams, D.G.; Coffee, R.L., Jr.; Zhang, H.; Pelech, S.; Strack, S.; Wadzinski, B.E. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J. Biol. Chem. 2005, 280, 42644–42654.
- Molkentin, J.D. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475.
This entry is offline, you can click here to edit this entry!