Astrocytes are essential players of brain circuit development and homeostasis controlling many aspects of synapse formation, function, plasticity and elimination both during development and adulthood. Accordingly, alterations in astrocyte morphogenesis and physiology may severely affect proper brain development, causing neurological or neuropsychiatric conditions. Recent findings revealed a huge astrocyte heterogeneity among different brain areas, which is likely at the basis of the different synaptogenic potential of these cells in selected brain regions.
- astrocyte factors
- astrocyte diversity
- synaptogenesis
- synaptic pruning
- synaptic plasticity
1.Introduction
The synaptogenic role of astrocytes was initially discovered using a purified retinal ganglion cell (RGC) culture system. RGC neurons grown in the absence of astroglia form very few synapses. However, synapse formation is increased upon addition of astrocyte-conditioned media (ACM)[1]. Since then, a large number of studies demonstrated that astrocytes are essential players in promoting synaptogenesis, particularly during brain development, and provided evidence for many astrocyte-secreted factors, including proteins, lipids, and small molecules that control different aspects of excitatory and inhibitory synapse formation and maturation. During the last two decades the astrocyte secretome, i.e. entire set of secreted proteins according to the definition coined by Tjalsma et al., in 2000[2][3], has been extensively analyzed in search of synapse-promoting factors. The full list of astrocytes’ molecules involved in synaptogenesis is reported in Table 1 and has been extensively reviewed in the last years, (see also [4][5][6][7]). Here we focus on three classes of well-known synaptogenic factors, which play key roles in early synaptogenesis through recently discovered molecular mechanisms.
2. Thrombospondins and Pentraxin 3.
In the first two weeks of postnatal mouse development, which correspond to a period of massive synaptogenesis in most brain areas, astrocyte-derived thrombospondin 1 and 2 (TSP1, TSP2) are highly expressed and initiate formation of synapses[8]. This process occurs through TSP binding membrane proteins, including the Ca2+ channel subunit α2δ-1[9] or the postsynaptic cell adhesion molecule Neuroligin1[10]. It has been demonstrated that TSPs induce the formation of ultrastructurally normal synaptic contacts, which are presynaptically active with cycling synaptic vesicles. Remarkably, they result postsynaptically silent because of the lack of functional 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA) receptors[8].
An elegant study by Risher and colleagues recently demonstrated that in the cerebral cortex TSP–α2δ-1 interaction controls synaptogenesis by acting postsynaptically via Rac1[11]. Indeed, they found that the synaptogenic function of α2δ-1 is cell autonomous to neurons, requiring the postsynaptic expression of α2δ-1. Upon TSP–α2δ1 interaction at the postsinaptic level, the guanine nucleotide exchange factors (GEFs), Kalirin-7 and β-Pix/Cool-1 β, activate Rac1 pathway and promote the remodeling of actin cytoskeleton at the nascent synaptic contact[11]. TSP1 and TSP2 display a developmentally regulated pattern of expression and are downregulated around the second and third weeks of postnatal development[8].It has been known for a long time that the activity of TSPs is fundamental to promote the formation of early synapses, which are silent. Which is/are the factors involved in the activation of these first synapses was not known until very recently. Indeed, it has been recently demonstrated that concomitantly to TSP secretion, astrocytes release the innate immune molecule pentraxin3 (PTX3), which promotes the functional maturation of excitatory synapses formed during the first wave of synaptogenesis by inducing AMPA receptors clustering at the synapse[12].
This occurs through a process involving the key PTX3 binding partner, TSG6, the remodeling of the perineural extracellular matrix surrounding synaptic contacts and requires β1-integrins. Quite unexpectedly, PTX3 deficient mice display weaker excitatory synapses in the hippocampus not only at young ages but also at P30, indicating that lack of endogenous PTX3 results in synapse defects, which apparently cannot be rescued by other astrocyte-derived factors expressed at later developmental stages[13][14][15], see next paragraphs. Notably, PTX3 is able to interact with TSP1 and 2, but not with TSP4. Upon binding to TSP1, PTX3 activity is inhibited, thus representing an additional mechanism of control of the process[12]. Notably, PTX3 and TSP1 display a spatially and temporally overlapped expression also in human brain, being higher in the astrocytes of foetal cerebral cortex[16]. The relative amount of the two molecules could therefore be crucial to set the proper balance between synaptic growth and synapse maturation during the period of early synaptogenesis. Both common and rare variants of the gene encoding for TSP1, THBS1, have been found to be associated with Autism Spectrum Disorders (ASD) in a cohort of 313 patients by Sanger sequencing[17]. Furthermore single nucleotide polymorphisms (SNPs) of PTX3 gene have been reported to result in PTX3 deficiency in humans and are associated with changed susceptibility to infections and altered inflammatory response[18][19][20]. Whether these PTX3 variants might be linked to altered synaptogenesis under physiological conditions or in response to an inflammatory insult occurring during brain development is presently not known.
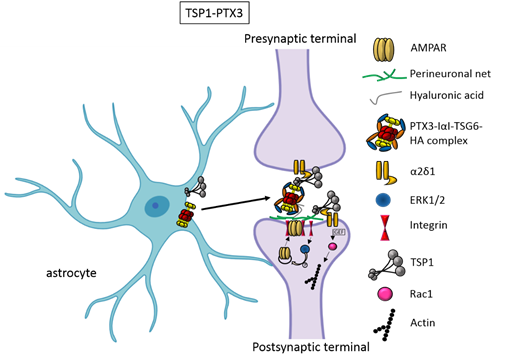
Figure 1. TSP1 and PTX3 cooperate to promote early formation of functional excitatory synapses. TSP1 binds to the α2δ1 receptor on the presynaptic terminals stimulating an increase of structurally normal but silent synapses and to the α2δ1 receptor at the postsynaptic site activating Rac1 and stimulating actin remodelling to promote spinogenesis. PTX3 promotes the functional maturation of these synapses by recruiting AMPA receptors at the synapse.
3. Hevin and SPARC.
Besides TSPs, two members of the secreted protein acidic, enriched in cysteine (SPARC) family proteins, Hevin (also known as SPARC-like1/SPARCL1) and SPARC were recognised as astrocyte-secreted factors that control synapse formation between cultured RGCs. Similar to TSPs, treatment of RGCs with purified Hevin is sufficient to induce formation of structurally mature, but postsynaptically silent synapses[15]. Analyses of developing visual cortices in Hevin-null mice revealed fewer thalamocortical synapses and spines displaying features of immaturity, indicating that the protein is required for proper development of the thalamocortical synaptic connectivity[21]. Astrocyte-derived Hevin has been demonstrated to serve as a bridge for the non-interacting isoforms of neurexin-1alpha and neuroligin-1B at the level of thalamocortical connections in the developing visual cortex[22]. By contrast, SPARC antagonizes the synaptogenic action of Hevin through a dominant-negative action, most likely by interfering with the ability of Hevin to bridge neurexin-1alpha and neuroligin-1B[15]. In addition, SPARC is able to trigger a cell-autonomous programme of synapse elimination[23]. In the developing mouse brain, Hevin and SPARC are expressed at high levels in astrocytes during the second and third weeks, which coincide with periods of synapse stabilization and synaptic refinement. Of note, SPARCL1 has been recently identified as part of a network of genes linked to neuronal damage in the preclinical stages of Alzheimer’s disease (AD)[25]. In particular, SPARCL1 variants that correlate with lower gene expression levels in brain are associated with accelerated cognitive decline during preclinical AD.
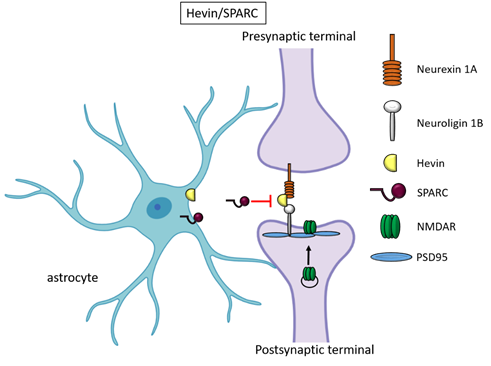
Figure 2. Hevin and SPARC. Astrocyte-secreted Hevin bridges presynaptic neurexin and postsynaptic neuroligin to favour the recruitment of PSD95 and NMDAR subunits at the synapse. SPARC on the contrary antagonizes Hevin effect with a still unknown mechanism.
4. Glypicans and neuronal pentraxins.
Recently, astrocyte-secreted glypicans 4 and 6 have been discovered by using the same experimental setting (i.e. RGC cultures and ACM) that led to the discovery of thrombospondins. Glypicans are expressed at later stages of development (second and third postnatal weeks). Exposure of RGC cultures to glypicans 4 or 6 resulted in enhanced formation of active excitatory synapses containing GluA1 receptors at difference to the action of TSPs [13]. GPC4 deficient mice show defective excitatory synapses in the hippocampus[13] and alteration in social behaviour which resemble ASD core symptoms and that can be linked with loss of GluA1[26]. The ability of glypican 4 to induce active synapses involves neuronal pentraxin 1 (NP1) which is released from presynaptic terminals by signaling through presynaptic protein tyrosine phosphatase receptor delta (PTPRδ). NP1 then stimulates AMPA receptors clustering on the postsynaptic dendrite thus allowing the assembly of functional synapses[14].
Neuronal pentraxins - the membrane-bound "neuronal pentraxin receptor" (NPR) and the secreted proteins NP1 and NARP (i.e., NP2) - are expressed by neurons and promote AMPA clustering at excitatory synapses by direct binding of the N- terminal domain of the receptor[27][28][29][30][31][32][33]. Recently it has been demonstrated that knockdown of NPR in hippocampal neurons dramatically decreases assembly and function of both excitatory and inhibitory postsynaptic specializations[34]. Evidence showed that NPR recruits and stabilizes NP1 and NARP on the presynaptic plasma membrane suggesting that neuronal pentraxins act as trans-synaptic organizers of both excitatory and inhibitory synapses. Diseases associated with GPC4 include Simpson-Golabi-Behmel Syndrome which in some cases results in mild to severe intellectual disability[35].
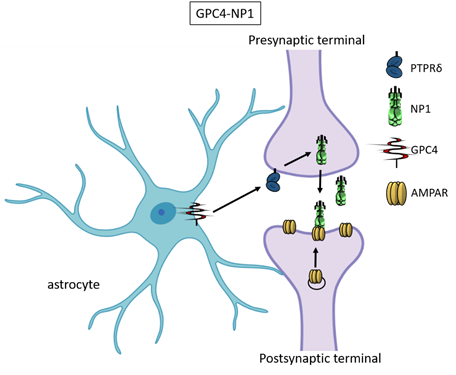
Figure 3. Glypican 4 and Neuronal Pentraxin 1. Astrocyte-derived Glypican 4 induces NP1 release from neurons through the PTPRδ receptor. NP1 stimulates AMPA receptors clustering on the postsynaptic terminal making the synapse functional.
This entry is adapted from the peer-reviewed paper 10.3390/cells9102173
References
- Ullian, E.M., et al., Control of synapse number by glia. Science, 2001. 291(5504): p. 657-61.
- Tjalsma, H., et al., Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev, 2000. 64(3): p. 515-47.
- Tjalsma, H., et al., Proteomics of protein secretion by Bacillus subtilis: separating the "secrets" of the secretome. Microbiol Mol Biol Rev, 2004. 68(2): p. 207-33.
- Chung, W.S., N.J. Allen, and C. Eroglu, Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb Perspect Biol, 2015. 7(9): p. a020370.
- Allen, N.J. and C. Eroglu, Cell Biology of Astrocyte-Synapse Interactions. Neuron, 2017. 96(3): p. 697-708.
- Bosworth, A.P. and N.J. Allen, The diverse actions of astrocytes during synaptic development. Curr Opin Neurobiol, 2017. 47: p. 38-43.
- Baldwin, K.T. and C. Eroglu, Molecular mechanisms of astrocyte-induced synaptogenesis. Curr Opin Neurobiol, 2017. 45: p. 113-120.
- Christopherson, K.S., et al., Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell, 2005. 120(3): p. 421-33.
- Eroglu, C., et al., Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell, 2009. 139(2): p. 380-92.
- Xu, J., N. Xiao, and J. Xia, Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nat Neurosci, 2010. 13(1): p. 22-4.
- Risher, W.C., et al., Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol, 2018. 217(10): p. 3747-3765.
- Fossati, G., et al., Pentraxin 3 regulates synaptic function by inducing AMPA receptor clustering via ECM remodeling and beta1-integrin. EMBO J, 2019. 38(1).
- Allen, N.J., et al., Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature, 2012. 486(7403): p. 410-4.
- Farhy-Tselnicker, I., et al., Astrocyte-Secreted Glypican 4 Regulates Release of Neuronal Pentraxin 1 from Axons to Induce Functional Synapse Formation. Neuron, 2017. 96(2): p. 428-445 e13.
- Kucukdereli, H., et al., Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A, 2011. 108(32): p. E440-9.
- Zhang, S., et al., Constitutive expression of pentraxin 3 (PTX3) protein by human amniotic membrane cells leads to formation of the heavy chain (HC)-hyaluronan (HA)-PTX3 complex. J Biol Chem, 2014. 289(19): p. 13531-42.
- Lu, L., et al., Common and rare variants of the THBS1 gene associated with the risk for autism. Psychiatr Genet, 2014. 24(6): p. 235-40.
- Barbati, E., et al., Influence of pentraxin 3 (PTX3) genetic variants on myocardial infarction risk and PTX3 plasma levels. PLoS One, 2012. 7(12): p. e53030.
- Cunha, C., O. Kurzai, and A. Carvalho, PTX3 deficiency and aspergillosis. N Engl J Med, 2014. 370(17): p. 1666-7.
- Bonacina, F., et al., Pentraxin 3 deficiency protects from the metabolic inflammation associated to diet-induced obesity. Cardiovasc Res, 2019. 115(13): p. 1861-1872.
- Risher, W.C., et al., Astrocytes refine cortical connectivity at dendritic spines. Elife, 2014. 3.
- Singh, S.K., et al., Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1alpha and NL1 via Hevin. Cell, 2016. 164(1-2): p. 183-196.
- Lopez-Murcia, F.J., B. Terni, and A. Llobet, SPARC triggers a cell-autonomous program of synapse elimination. Proc Natl Acad Sci U S A, 2015. 112(43): p. 13366-71.
- Clarke, L.E. and B.A. Barres, Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci, 2013. 14(5): p. 311-21.
- Seddighi, S., et al., SPARCL1 Accelerates Symptom Onset in Alzheimer's Disease and Influences Brain Structure and Function During Aging. J Alzheimers Dis, 2018. 61(1): p. 401-414.
- Dowling, C. and N.J. Allen, Mice Lacking Glypican 4 Display Juvenile Hyperactivity and Adult Social Interaction Deficits. Brain Plast, 2018. 4(2): p. 197-209.
- O'Brien, R.J., et al., Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron, 1999. 23(2): p. 309-23.
- O'Brien, R., et al., Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultured spinal neurons. J Neurosci, 2002. 22(11): p. 4487-98.
- Xu, D., et al., Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron, 2003. 39(3): p. 513-28.
- Sia, G.M., et al., Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron, 2007. 55(1): p. 87-102.
- Chang, M.C., et al., Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat Neurosci, 2010. 13(9): p. 1090-7.
- Gu, Y., et al., Obligatory role for the immediate early gene NARP in critical period plasticity. Neuron, 2013. 79(2): p. 335-46.
- Pelkey, K.A., et al., Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron, 2015. 85(6): p. 1257-72.
- Lee, S.J., et al., Presynaptic Neuronal Pentraxin Receptor Organizes Excitatory and Inhibitory Synapses. J Neurosci, 2017. 37(5): p. 1062-1080.
- Tenorio, J., et al., Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis, 2014. 9: p. 138.