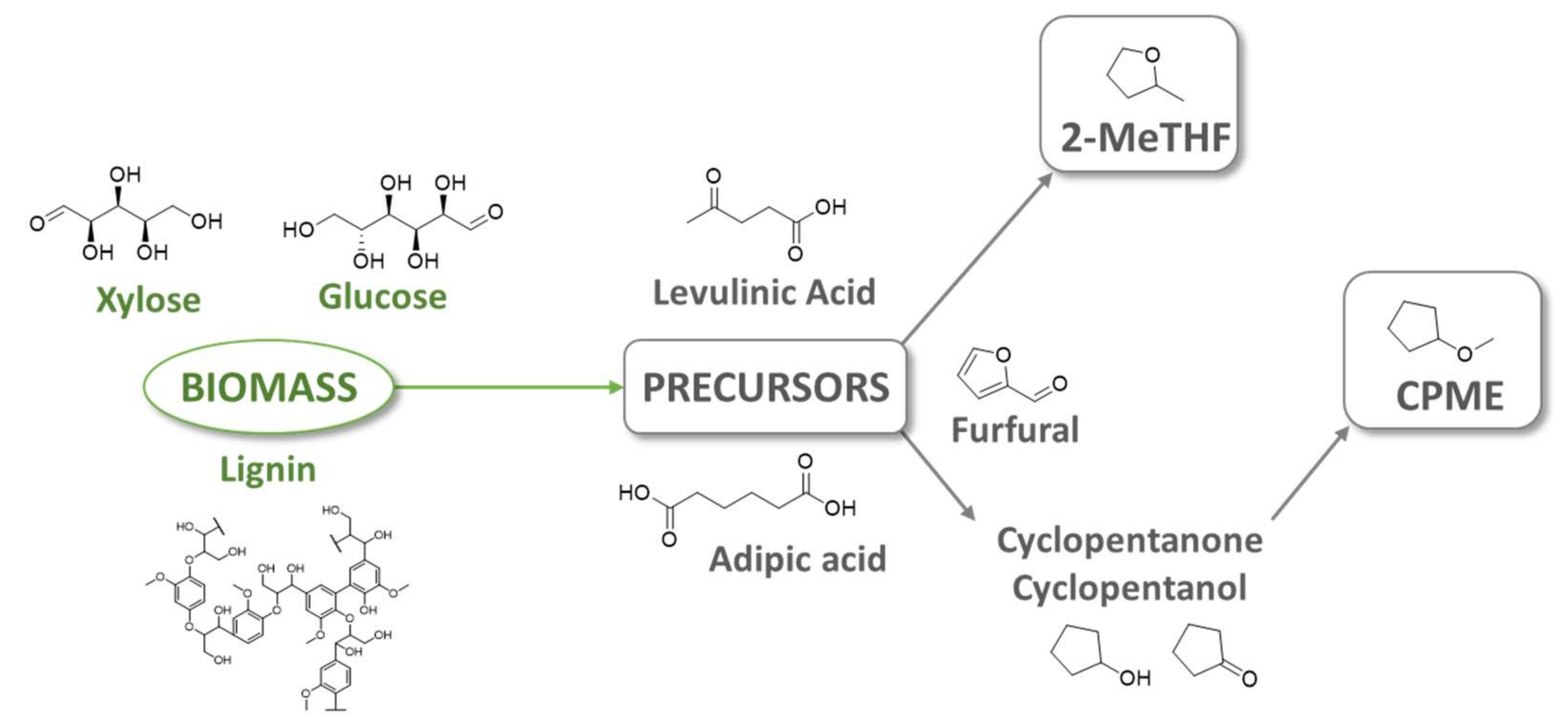
Biocatalysis can be defined in a broad sense as the mediation of chemical reactions by means of biological systems, including isolated enzymes, whole cells or cell-free extracts. In some circumstances, the aqueous buffer medium normally employed in biocatalytic procedures is not the best option to develop these processes, due to solubility and/or inhibition issues, requiring biocatalyzed redox procedures to circumvent these drawbacks, by developing novel green non-conventional media, including the use of biobased solvents, reactions conducted in neat conditions and the application of neoteric solvents such as deep eutectic solvents.
- biocatalysis
- deep eutectic solvents
- biobased solvents
- neat conditions
1. Introduction
2. Biocatalyzed Redox Processes at Neat Conditions
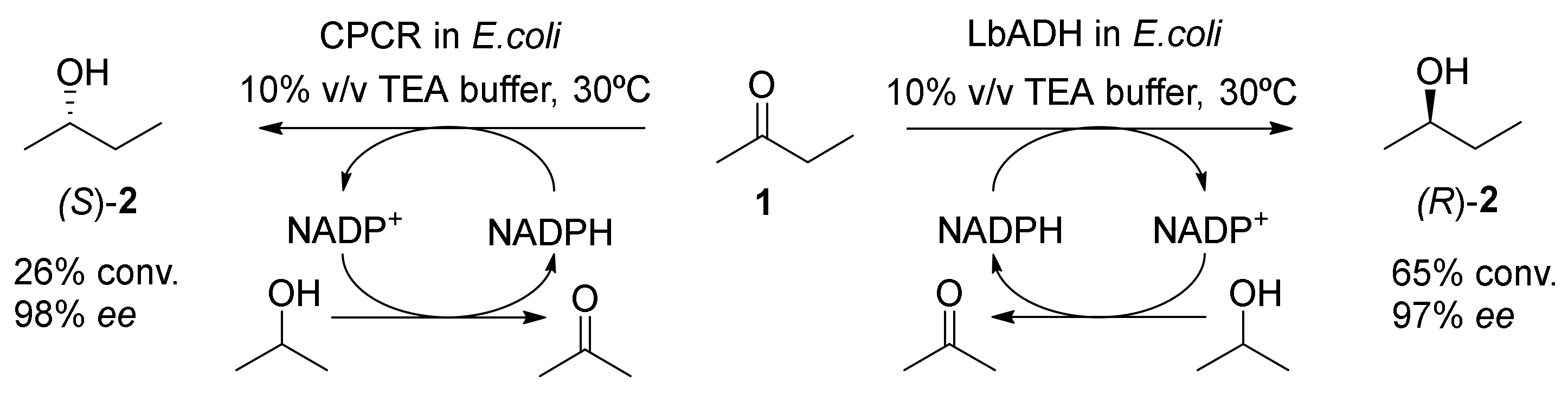
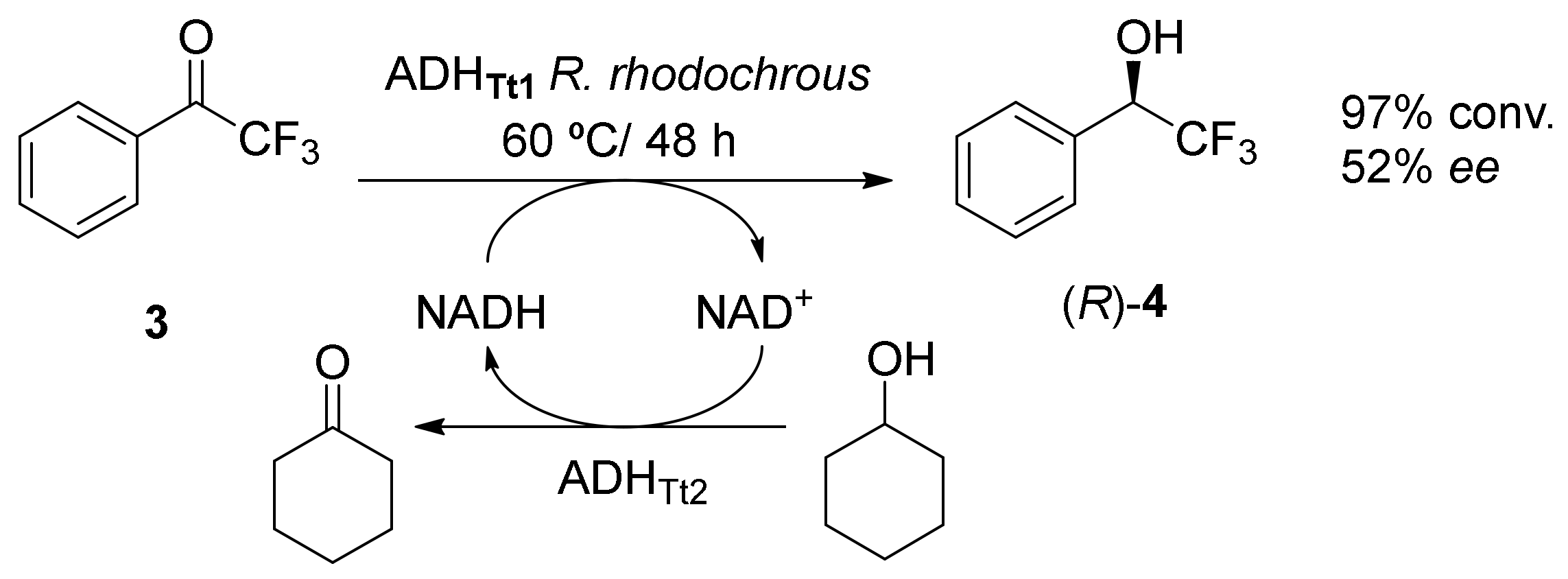
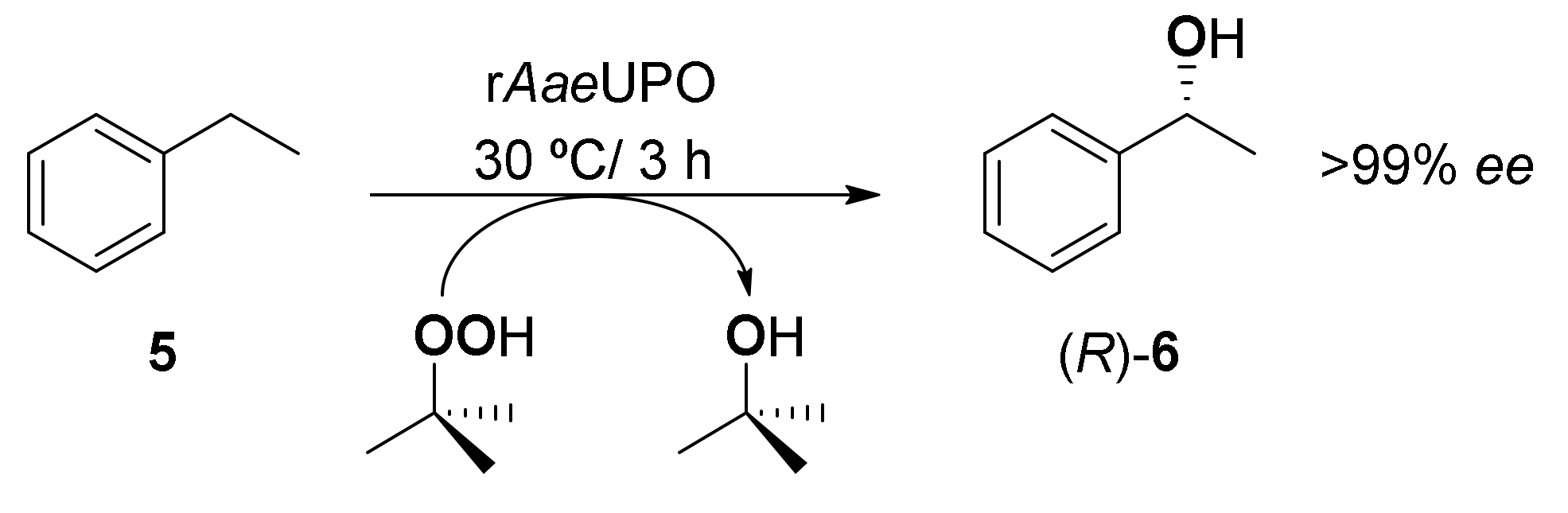
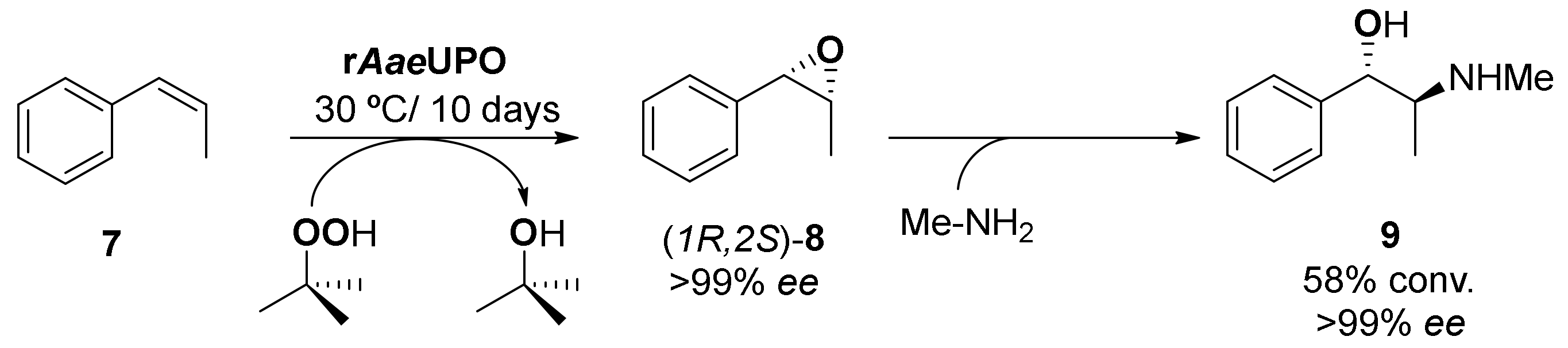
3. Application of Bio-Based Solvents in Redox Reactions Catalyzed by Enzymes
|
Biocatalyst |
Solvent (% v/v) |
Yield (%) |
ee (%) |
Reference |
|---|---|---|---|---|
|
Enoate reductases |
CPME 20% v/v |
<10 |
0–99 |
[53] |
|
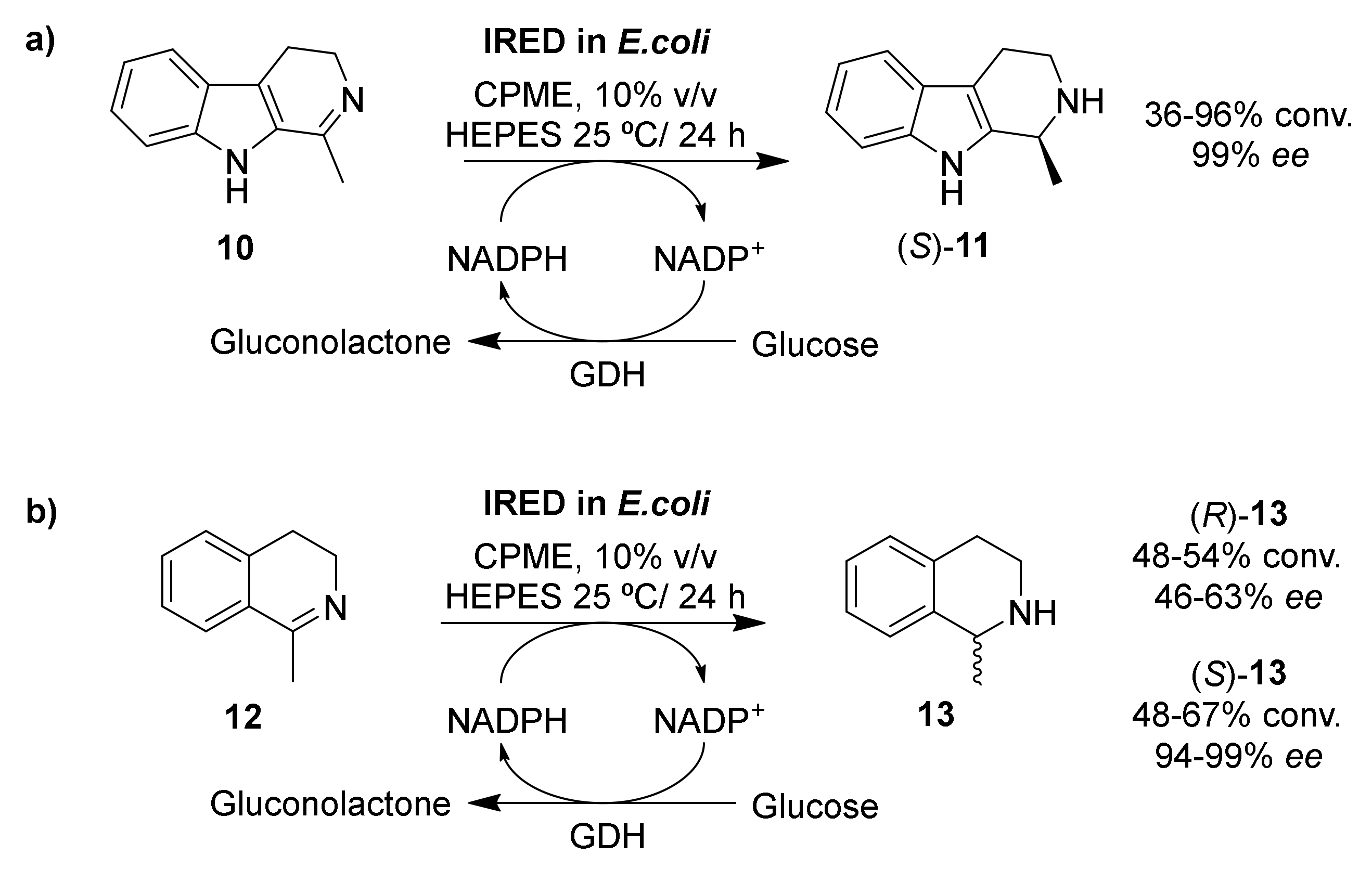
Imine reductases |
CPME 90% v/v |
36–96 |
46–99 |
[58] |
|
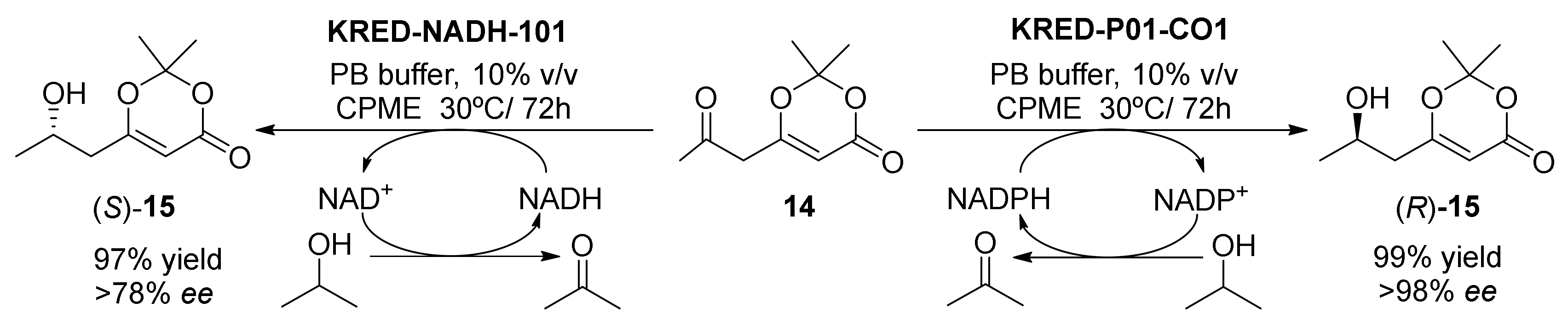
ADH |
CPME 10% v/v |
>90 |
>90 |
[59] |
|
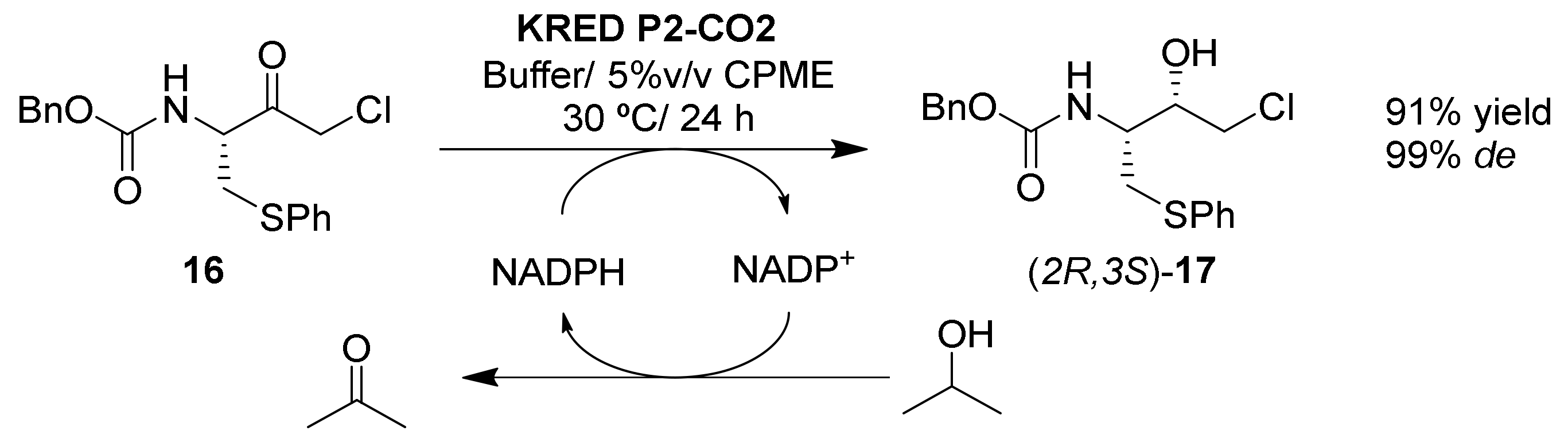
ADH |
CPME 5% v/v |
99 |
99 |
[60] |
|
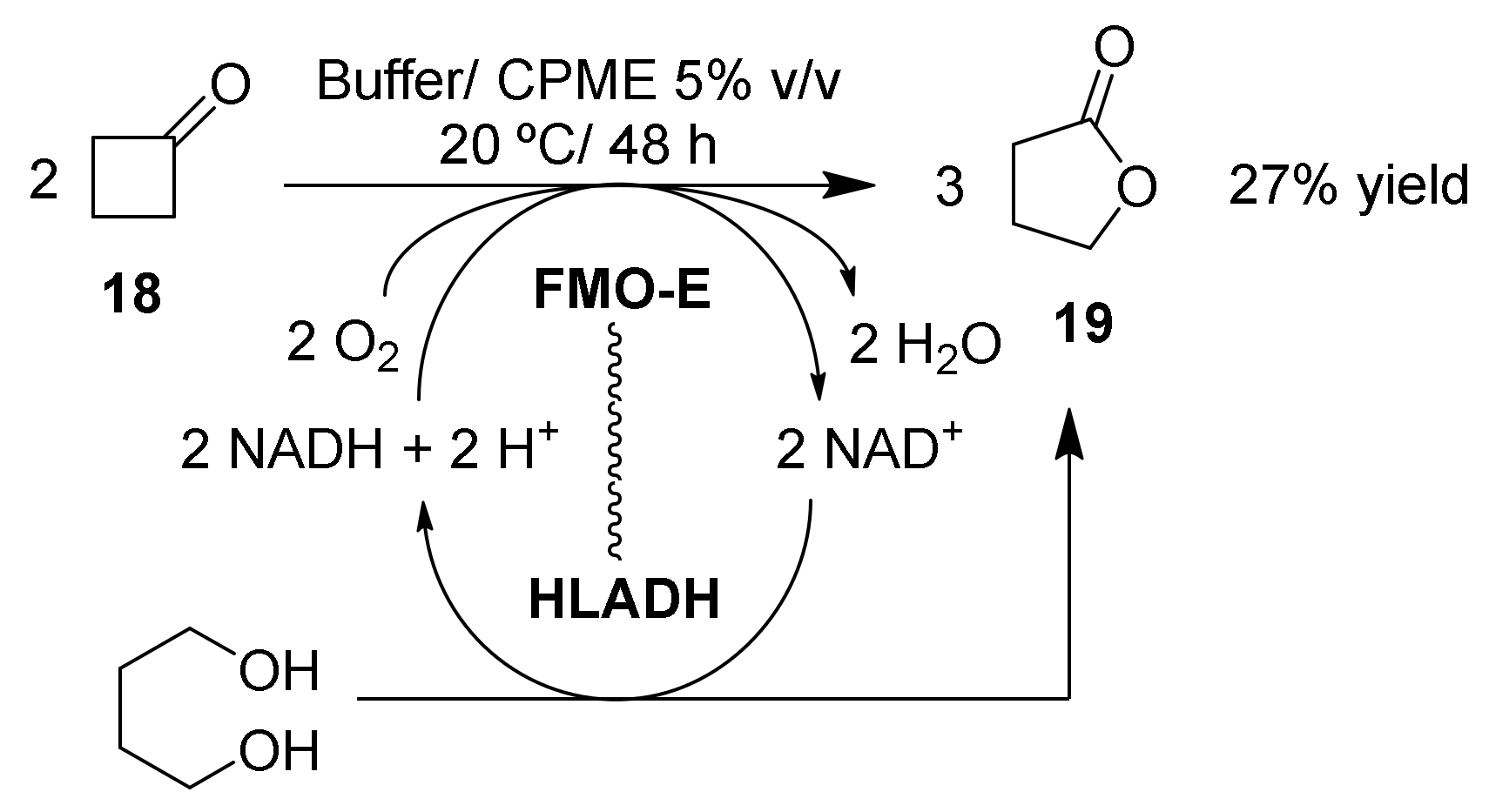
Monooxygenase + ADH |
CPME 5% v/v |
27 |
- |
[61] |
|
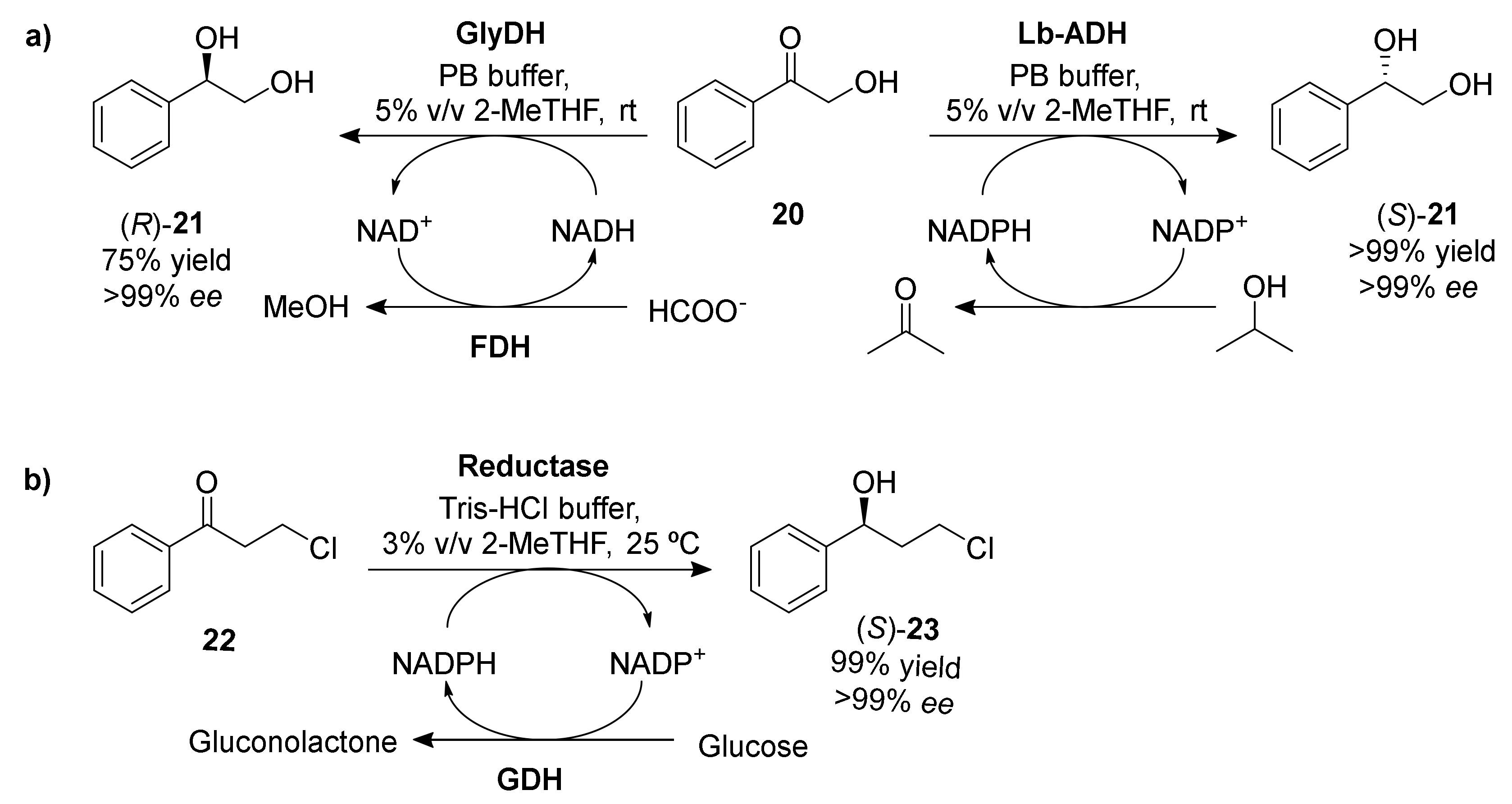
Alcohol and glycerol dehydrogenases |
2-MeTHF 5% v/v |
75–99 |
>99 |
[64] |
|
Saccharomyces cerevisiae YOL151W Reductase |
2-MeTHF 3% v/v |
99 |
>99 |
[65] |
4. Deep-Eutectic Solvents in Bioreductions/Biooxidations
This entry is adapted from the peer-reviewed paper 10.3390/molecules25133016
References
- Hudlicky, T.; Reed, J.W. Applications of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 38, 3117–3132.
- Sheldon, R.A.; Brady, D. Broadening the scope of biocatalysis in sustainable organic synthesis. ChemSusChem 2019, 12, 2859–2881.
- Sheldon, R.A.; Woodley, J.M. Role of biocatalysis in sustainable chemistry. Chem. Rev. 2018, 118, 801–838.
- Dominguez de María, P.; de Gonzalo, G.; Alcántara, A.R. Biocatalysis as useful tool in asymmetric synthesis: An assessment of recently granted patents (2014–2019). Catalysts 2019, 9, 802.
- Sheldon, R.A.; Brady, D. The limits to biocatalysis: Pushing the envelope. Chem. Commun. 2018, 54, 6088–6144.
- Domínguez de María, P.; de Gonzalo, G. (Eds.) Biocatalysis: An Industrial Perspective, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 2018.
- Patel, R.N. Biocatalysis for the synthesis of pharmaceuticals. Bioorganic Med. Chem. 2018, 26, 1252–1274.
- Hollmann, F.; Opperman, D.J.; Paul, C.E. Enzymatic reductions: A chemist’s perspective. Angew. Chem. Int. Ed. 2020.
- De Gonzalo, G.; Lavandera, I. Recent advances in selective biocatalytic (hydrogen transfer) reductions. In Homogeneous Hydrogenation with Non-Precious-Catalysts, 1st ed.; Teichert, J.F., Ed.; Wiley-VCH: Weinheim, Germany, 2020; pp. 227–259.
- Dong, J.J.; Fernández-Fueyo, E.; Hollmann, F.; Paul, C.E.; Pesic, M.; Schmidt, S.; Wang, Y.H.; Younes, S.; Zhang, W.Y. Biocatalytic oxidation reactions: A chemist’s perspective. Angew. Chem. Int. Ed. 2018, 57, 9238–9261.
- Holtmann, D.; Fraaije, M.W.; Arends, I.W.C.E.; Opperman, D.J.; Hollmann, F. The taming of oxygen: Biocatalytic oxyfunctionalizations. Chem. Commun. 2014, 50, 13180–13200.
- Domínguez de María, P.; Hollmann, F. On the (un)greenness of biocatalysis: Some challenging figures and some promising options. Front. Microbiol. 2016, 6, 1257.
- Zaks, A.; Klibanov, A.M. Enzymatic catalysis in nonaqueous solvents. J. Biol. Chem. 1988, 263, 3194–3201.
- Zaks, A.; Klibanov, A.M. Enzyme-catalyzed processes in organic solvents. Proc. Natl. Acad. Sci. USA 1985, 82, 3192–3196.
- Carrea, G.; Riva, S. Properties and synthetic applications of enzymes in organic solvents. Angew. Chem. Int. Ed. Engl. 2000, 39, 2226–2254.
- Hernáiz, M.J.; Alcántara, A.R.; García, J.I.; Sinisterra, J.V. Applied biotransformations in green solvents. Chem. Eur. J. 2010, 16, 9422–9437.
- Sheldon, R.A. The E factor 25 years on: The rise of green chemistry and sustainability. Green Chem. 2017, 19, 18–43.
- Pace, V.; Hoyos, P.; Castoldi, L.; Domínguez de María, P.; Alcántara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A biomass-derived solvent with broad application in organic chemistry. ChemSusChem 2012, 5, 1369–1379.
- De Gonzalo, G.; Alcántara, A.R.; Domínguez de María, P. Cyclopentyl methyl ether (CPME): A versatile eco-friendly solvent for applications in biotechnology and biorefineries. ChemSusChem 2019, 12, 2083–2097.
- Clarke, C.J.; Tu, W.C.; Levers, O.; Bröhl, A.; Hallet, J.P. Green and sustainable solvents in chemical processes. Chem. Rev. 2018, 118, 747–800.
- Calvo-Flores, F.G.; Monteagudo-Arrebola, M.J.; Dobado, J.A.; Isac-García, J. Green and bio-based solvents. Top. Curr. Chem. 2018, 376, 18.
- Guajardo, N.; Domínguez de María, P. Assessing biocatalysis using dihydrolevoglucosenone (Cyrene™) as versatile bio-based (co)solvent. Mol. Catal. 2020, 485, 110813.
- Hoang, H.N.; Matsuda, T. Expanding substrate scope of lipase-catalyzed transesterification by the utilization of liquid carbon dioxide. Tetrahedron 2016, 72, 7229–7234.
- Matsuda, T. Recent progress in biocatalysis using supercritical carbon dioxide. J. Biosci. Bioeng. 2013, 115, 233–241.
- Cantone, S.; Hanefeld, U.; Basso, A. Biocatalysis in non-conventional media—Ionic liquids, supercritical fluids and the gas phase. Green Chem. 2007, 9, 954–971.
- Hoang, H.N.; Nagashima, Y.; Mori, S.; Kagechika, H.; Matsuda, T. CO2-expanded bio-based liquids as novel solvents for enantioselective biocatalysis. Tetrahedron 2017, 73, 2984–2989.
- Itoh, T. Ionic liquids as tool to improve enzymatic organic synthesis. Chem. Rev. 2017, 117, 10567–11607.
- Lozano, P.; Alvárez, E.; Bernal, J.M.; Nieto, S.; Gómez, C.; Sánchez-Gómez, G. Ionic liquids for clean biocatalytic processes. Curr. Green Chem. 2017, 4, 116–129.
- Domínguez de María, P. (Ed.) Ionic Liquids in Biotransformations and Organocatalysis, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2012.
- Domínguez de María, P.; Maugeri, Z. Ionic liquids in biotransformations: From proof of concept to emerging deep-eutectic-solvents. Curr. Opin. Chem. Biol. 2011, 15, 220–225.
- Mbous, Y.P.; Hayyan, M.; Hayyan, A.; Wong, W.F.; Hashim, M.A.; Looi, C.Y. Applications of deep eutectic solvents in biotechnology and bioengineering—Promises and challenges. Biotechnol. Adv. 2017, 35, 105–134.
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep eutectic solvents (DESs) and their applications. Chem. Rev. 2014, 114, 11060–11082.
- Zhang, Q.H.; Vigier, K.D.; Royer, S.; Jerome, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146.
- Heeres, A.; Vanbroekhoven, K.; Van Hecke, W. Solvent-free lipase-catalyzed production of (meth) acrylate monomers: Experimental results and kinetic modeling. Biochem. Eng. J. 2019, 142, 162–169.
- Wunschik, D.S.; Ingenbosch, K.N.; Zähres, M.; Horst, J.; Mayer, C.; Jäger, M.; Strehmel, V.; Dornbusch, M.; Hoffmann-Jacobsen, K. Biocatalytic and solvent-free synthesis of a bio-based biscyclocarbonate. Green Chem. 2018, 20, 4738–4745.
- Böhmer, W.; Koenekoop, L.; Simon, T.; Mutti, F. Parallel Interconnected Kinetic Asymmetric Transformation (PIKAT) with an immobilized ω-transaminase in neat organic solvent. Molecules 2020, 25, 2140.
- Hobbs, H.R.; Thomas, N.R. Biocatalysis in supercritical fluids, fluorous solvents and under solvent-free conditions. Chem. Rev. 2007, 107, 2786–2820.
- Kara, S.; Spickermann, D.; Weckbecker, A.; Leggewie, C.; Arends, I.W.C.E.; Hollmann, F. Bioreductions catalyzed by an alcohol dehydrogenase in non-aqueous media. ChemCatChem 2014, 6, 973–976.
- Zheng, Y.-G.; Yin, H.-H.; Yu, D.-F.; Chen, X.; Tang, X.-L.; Zhang, X.-J.; Xue, Y.-P.; Wang, Y.-J.; Liu, Z.-Q. Recent advances in biotechnological applications of alcohol dehydrogenases. Appl. Microbiol. Biotechnol. 2017, 101, 987–1001.
- Moody, T.S.; Rozzell, J.D. Modern biocatalytic ketone reduction. In Organic Synthesis Using Biocatalysts; Goswami, A., Stewart, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 149–186.
- Nealon, C.M.; Musa, M.M.; Patel, J.M.; Phillips, R.S. Controlling substrate specificity and stereospecificity of alcohol dehydrogenases. ACS Catal. 2015, 5, 2100–2114.
- De Gonzalo, G.; Lavandera, I.; Faber, K.; Kroutil, W. Enzymatic reduction of ketones in “micro-aqueous” media catalyzed by ADH-a from Rhodococcus ruber. Org. Lett. 2007, 9, 2163–2166.
- Jakoblinnert, A.; Mladenov, R.; Paul, A.; Sibilla, F.; Schwaneberg, U.; Ansorge-Schumacher, M.B.; Domínguez de María, P. Asymmetric reduction of ketones with recombinant E. coli whole cells in neat substrates. Chem. Commun. 2011, 47, 12230–12232.
- Erdmann, V.; Mackfeld, U.; Rother, D.; Jakoblinnert, A. Enantioselective, continuous (R)- and (S)-2-butanol synthesis: Achieving high space-time yields with recombinant E. coli cells in a micro-aqueous, solvent-free reaction system. J. Biotechnol. 2014, 191, 106–112.
- Hibino, A.; Ohtake, H. Use of hydrophobic bacterium Rhodococcus rhodochrous NBRC15564 expressed thermophilic alcohol dehydrogenases as whole-cell catalyst in solvent-free organic media. Process Biochem. 2013, 48, 838–843.
- Fernández-Fueyo, E.; Ni, Y.; Gomez Baraibar, A.; Alcalde, M.; van Langen, L.M.; Hollmann, F. Towards preparative peroxygenase-catalyzed oxyfunctionalization reactions in organic media. J. Mol. Catal. B Enzym. 2016, 134, 347–352.
- Rauch, M.C.R.; Tieves, F.; Paul, C.E.; Arends, I.W.C.E.; Alcalde, M.; Hollmann, F. Peroxygenase-catalysed epoxidation of styrene derivatives in neat reaction media. ChemCatChem 2019, 11, 4519–4523.
- Hofrichter, M.; Kellner, H.; Herzog, R.; Karich, A.; Liers, C.; Scheibner, K.; Wambui, V.; Ullrich, R. Fungal peroxygenases: A phylogenetically old superfamily of heme enzymes with promiscuity for oxygen transfer reaction. In Grand Challenges in Fungal Biotechnology; Nevalainen, H., Ed.; Springer: Cham, Switzerland, 2020.
- Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F. Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges? Curr. Opin. Chem. Biol. 2017, 37, 1–9.
- Tieves, F.; Willot, S.J.P.; van Schi, M.M.C.H.; Rauch, M.C.R.; Younes, S.H.H.; Zhang, W.; Dong, J.; Gomez de Santos, P.; Robbins, J.M.; Bommarius, B.; et al. Formate Oxidase (FOx) from Aspergillus oryzae: One catalyst enables diverse H2O2-dependent biocatalytic oxidation reactions. Angew. Chem. Int. Ed. 2019, 58, 7873–7877.
- Freakley, S.J.; Kochius, S.; van Marwijk, J.; Fenner, C.; Lewis, R.J.; Baldenius, K.; Marais, S.S.; Opperman, D.J.; Harrison, S.T.L.; Alcalde, M.; et al. A chemo-enzymatic oxidation cascade to activate C–H bonds with in situ generated H2O2. Nat. Commun. 2019, 10, 4178.
- Azzena, U.; Carraro, M.; Pisano, L.; Monticelli, S.; Bartolotta, R.; Pace, V. Cyclopentyl methyl ether: An elective ecofriendly solvent in classical and modern chemistry. ChemSusChem 2019, 12, 40–70.
- Reß, T.; Hummel, W.; Hanlon, S.P.; Iding, H.; Gröger, H. The organic–synthetic potential of recombinant ene reductases: Substrate-scope evaluation and process optimization. ChemCatChem 2015, 7, 1302–1311.
- Toogood, H.S.; Scrutton, N.S. Discovery, characterization, engineering, and applications of ene-reductases for industrial biocatalysis. ACS Catal. 2018, 8, 3532–3549.
- Winkler, C.K.; Faber, K.; Hall, M. Biocatalytic reduction of activated CC-bonds and beyond: Emerging trends. Curr. Opin. Chem. Biol. 2018, 43, 97–105.
- Mangas-Sanchez, J.; France, S.P.; Montgomery, S.L.; Aleku, G.A.; Man, H.; Sharma, M.; Ramsden, J.I.; Grogan, G.; Turner, N.J. Imine reductases (IREDs). Curr. Opin. Chem. Biol. 2017, 37, 19–25.
- Grogan, G.; Turner, N.J. InspIRED by nature: NADPH-dependent imine reductases (IREDs) as catalysts for the preparation of chiral amines. Chem. Eur. J. 2016, 22, 1900–1907.
- Maugeri, Z.; Rother, D. Application of imine reductases (IREDs) in micro-aqueous reaction systems. Adv. Synth. Catal. 2016, 358, 2745–2750.
- Betori, R.; Miller, E.R.; Scheidt, K.A. A biocatalytic route to highly enantioenriched β-hydroxydioxinones. Adv. Synth. Catal. 2017, 359, 1131–1137.
- Castoldi, L.; Ielo, L.; Hoyos, P.; Hernáiz, M.J.; De Luca, L.; Alcántara, A.R.; Holzer, W.; Pace, V. Merging litium carbenoid homologation and enzymatic reduction: A combinative approach to the HIV-protease inhibitor Nelfinavir. Tetrahedron 2018, 74, 2211–2217.
- Huang, L.; Aalbers, F.S.; Tang, W.; Röllig, R.; Fraaije, M.W.; Kara, S. Convergent cascade catalyzed by monooxygenase–alcohol dehydrogenase fusion applied in organic media. ChemBioChem 2019, 20, 1653–1658.
- Alcántara, A.R.; Domínguez de María, P. Recent advances on the use of 2-methyltetrahydrofuran (2-MeTHF) in biotransformations. Curr. Green Chem. 2018, 5, 85–102.
- Shanmuganathan, S.; Natalia, D.; van den Wittenboer, A.; Kohlmann, C.; Greiner, L.; Domínguez de María, P. Enzyme-catalyzed C–C bond formation using 2-methyltetrahydrofuran (2-MTHF) as (co)solvent: Efficient and bio-based alternative to DMSO and MTBE. Green Chem. 2010, 12, 2240–2245.
- Shanmuganathan, S.; Natalia, D.; Greiner, L.; Domínguez de María, P. Oxidation-hydroxymethylation-reduction: A one-pot three-step biocatalytic synthesis of optically active α-aryl vicinal diols. Green Chem. 2012, 14, 94–97.
- Tian, Y.; Ma, X.; Yang, M.; Wei, D.; Su, E. Synthesis of (S)-3-chloro-1-phenylpropanol by permeabilized recombinant Escherichia coli harboring Saccharomyces cerevisiae YOL151W reductase in 2-methyltetrahydrofuran cosolvent system. Catal. Commun. 2017, 97, 56–59.
- Alonso, D.A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I.M.; Ramon, D.J. Deep eutectic solvents: The organic reaction medium of the century. Eur. J. Org. Chem. 2016, 2016, 612–632.
- Liu, Y.; Friesen, J.B.; Lankin, J.B.; Chen, S.-N.; Pauli, G.F. Natural deep eutectic solvents: Properties, applications and perspectives. J. Nat. Prod. 2018, 81, 679–690.
- Gorke, J.T.; Srienc, F.; Kazlauskas, R.J. Hydrolase-catalyzed biotransformations in deep eutectic solvents. Chem. Commun. 2008, 10, 1235–1237.
- Domínguez de María, P.; Guajardo, N.; Kara, S. Enzyme catalysis: In DES, with DES, and in the presence of DES. In Deep Eutectic Solvents: Synthesis, Properties and Applications; Ramón, D.J., Guillena, G., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp. 257–272.
- Pätzold, M.; Siebenhaller, S.; Kara, S.; Liese, A.; Syldakt, C.; Holtmann, D. Deep eutectic solvents as efficient solvents in biocatalysis. Trends Biotechnol. 2018, 37, 943–959.
- Guajardo, N.; Müller, C.R.; Schrebler, R.; Carlesi, C.; Domínguez de Maria, P. Deep eutectic solvents for organocatalysis, biotransformations, and multistep organocatalyst/enzyme combinations. ChemCatChem 2016, 8, 1020–1027.
- Stepankova, V.; Vanacek, P.; Damborsky, J.; Chaloupkova, R. Comparison of catalysis by haloalkane dehalogenases in aqueous solutions of deep eutectic and organic solvents. Green Chem. 2014, 16, 2754–2761.
- Maugeri, Z.; Domínguez de María, P. Benzaldehyde lyase (BAL)-catalyzed enantioselective C-C bond formation in deep-eutectic-solvents–buffer mixtures. J. Mol. Catal. B Enzym. 2014, 107, 120–123.
- Gotor-Fernández, V.; Paul, C.E. Deep eutectic solvents for redox biocatalysis. J. Biotechnol. 2019, 293, 24–35.
- Hasani, F.Z.I.M.; Amzazi, S.; Lavandera, I. The versatile applications of DES and their influence on oxidoreductase-mediated transformations. Molecules 2019, 24, 2190.