Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Colorectal cancer is one of the most prevalent cancer types. Although there have been breakthroughs in its treatments, a better understanding of the molecular mechanisms and genetic involvement in colorectal cancer will have a substantial role in producing novel and targeted treatments with better safety profiles.
- Colorectal cancer
- Driver Genes
- APC
- TP53
- Protein kinases
1. Introduction
Cancer does not develop from a single gene defect in a similar way to how it occurs in other diseases such as cystic fibrosis or muscular dystrophy. Instead, cancer becomes invasive in the event that there are multiple cancer gene mutations where the safeguarding mechanisms could not protect the normal and healthy mammalian cells from their lethal effects. As a result, it is better to think of cancer genes that have been altered as contributing to, rather than causing, cancer [1]. The development of colorectal cancer involves a multiple step process incited by a distinctive genomic instability which encourages the cancerous cells to multiply, as well as increases the chances of cell survival.
Colorectal cancer has three recognized primary molecular groupings in terms of molecular genetics. The most prevalent one is the “chromosomal instable” group, which is defined by an accumulation of mutations in certain oncogenes and tumor suppressor genes. Chromosomal instability is the most common type of genomic instability in CRC. It is characterized by various changes in chromosomal copy number and structure. The normal activities of certain tumor-suppressor genes, such as APC, P53, and SMAD4, can be altered via a mechanism triggered by chromosomal instability which is responsible for the physical loss of a wild-type copy of these tumor suppressor genes. The second group is the CpG Island Methylation phenotype (CIMP), which is defined by DNA hypermethylation [2], as additional genes were discovered to be influenced by the process, revealing that some groupings of genes had consistently elevated methylation in particular tumors. This was proved statistically by demonstrating that the methylation of two distinct genes in a specific tumor type was associated in cases such as colorectal cancer [3].
The third group is the “microsatellite instable” (MSI) colorectal cancer thatis caused by DNA mismatch repair gene failure, resulting in genetic hypermutability. High MSI was found in 75% of this group, which is often linked with hypermethylation and MLH1 gene silence, whereas the remaining 25% had mutations in the mismatch-repair and polymerase (POLE) genes [4]. Generally, genomic instability can cause aggregation of mutations in genes that are responsible for normal cell regulation and growth, such as proto-oncogenes and tumor suppressor genes [5]. It can also derange the normal cell repair system, induce epigenetic changes in DNA, and produce non-functional proteins that could threaten the healthy cells. Notably, the significant types of genomic instability involved in the development of colorectal cancer are chromosomal instability but microsatellite stable and microsatellite instability (MSI) [6]. Markedly, MSI is often associated with the CpG island methylator phenotype and hypermutation, which is essentially found in the right colon [7]. Furthermore, parallel investigations revealed that the mismatch repair gene MLH1 was hypermethylated and silenced in these MSI-positive tumors. The fact that inhibiting methylation repaired the mismatch repair deficit in colon cancer cell lines supported the hypothesis that hypermethylation causes MSI through MLH1 silencing [3]. MSI affects the size of the mononucleotide or dinucleotide repeats, which are also known as microsatellites, existing all over the genome. It occurs when the strand slippage within the repetitive DNA sequence element failed to be repaired. Such instability resulting from the loss of mismatch-repair function of proteins in DNA can further contribute to the inactivation of the tumor suppression pathway [6].
A cancerous tumor can be characterized by low frequency of somatic mutations such as single nucleotide variants (SNVs), copy number aberrations (CNAs), structural variations, and indels. As indicated by the name, SNVs are aroused by a single nucleotide variant that occurred in one particular genetic position, while CNAs are the amplifications or deletions of copies of a DNA region at a larger scale. However, structural variation is used to describe an area of DNA that is 1 kb or bigger in size and can include inversions, balanced translocations, and genomic imbalances, which are also known as copy number variations. Insertions and deletions, called indels, are changes to the DNA sequence that result in the addition or deletion of one or more nucleotides [8]. Only a small percentage of all somatic changes, known as driver mutations, offer a selective advantage to cancer cells, whereas the vast majority of somatic mutations are passenger mutations that do not contribute to the illness [9]. Inter-tumor heterogeneity, where cancer genomes do not share a similar set of somatic mutations and most of the different metastatic tumors bear a different kind of mutation in the same patient, is the most remarkable trait of the cancer mutational landscape [10]. Besides, in less than 5% of all patients with a specific cancer type, a small number of gene mutations are found in a large portion of tumors and mostly are affected by SNVs or CNAs [11]. Inter-tumor heterogeneity impedes efforts to discover driver genes with driver mutations by recognizing commonly mutated genes that are mutated in a statistically high proportion of patients [12]. The nature of the driver mutations in targeting normal functional genes, groups of interacting proteins, as well as signaling and molecular pathways, is one of the causes of inter-tumor heterogeneity [13].
In silico techniques have long been considered crucial in the efforts of predicting inhibitors, new targets, and diagnostic tools for CRC treatment plans. Exploring binding pockets, residue interactions, and different virtual screening methods are approaches, among others, that were utilized to target CRC [14]. Gene-mutated CRC was targeted by topological in-silico simulations to predict the best treatment combinations that can be successful in clinically advanced conditions [15]. Furthermore, other tactics, such as the simulations that predict the interplay between tumor microenvironment components, could enhance or reduce immunotherapy success or failure [16], and the gut-on-chip model that delineates the molecular mechanism of symbiotic effects on CRC genes’ expression [17] are examples of significant accomplishments in this field. The use of computational methods has also proved a distinguished efficacy by analyzing cell surface proteins overexpression in predicting disease progression, diagnosis, and drug resistance in CRC [18]. MicroRNA was employed as a biomarker for CRC through its attachment to the predicted target gene. The molecular pathways and functional analysis of this non-coding RNA with its target macromolecules can predict CRC pathogenesis [19].
2. Driver Genes in CRC
Multistep tumorigenesis develops through the gradual collection and alterations of driver genes in colorectal cancer. Less than 1% of human genes can potentially turn into cancerous driver genes which are actively capable of controlling cell survival and fate, as well as affecting normal genome stability [10][20]. For a mature cell to become cancerous, it has to undergo phases of breakthrough, expansion, and invasion within 20 to 30 years, involving at least 2 to 3 driver gene mutations. It begins with the first driver mutation which minimally benefits the cell to survive and turns into a proliferating hyperplastic lesion. This could increase the risk of acquiring the second driver gene mutation and further leads to the third driver gene mutation as the cell gained autonomy and immortality, as well as the ability to self-renew. In the case when a third driver gene is involved, the tumor cell is upgraded to become invasive and metastatic. At this point, the malignant cells disseminate without the assistance of other driver mutations [21]. The International Cancer Genome Consortium (ICGC) platform shows the top 20 mutated genes in CRC such as APC, TP53, LRP1B, KRAS, and BRAF, which are significantly impacted by single somatic mutations that also have high functional impact as shown in Figure 1a. ICGC is a global platform that has compiled data on 670,946 unique somatic mutations and molecular profiles from 866 donors for CRC patients. These collected data are grouped into three CRC-related projects, namely, colon adenocarcinoma—TGCA, USA (COAD-US), non-Western colorectal cancer—China (COCA-CN), and rectum adenocarcinoma—USA (READ-US). In the same context, the Cancer Genome Atlas project profiled genomic changes in three cancer types; glioblastoma and ovarian carcinoma, in addition to colon and rectal cancer, among 20 different cancer types with a comprehensive molecular characterization for each one of them [7]. In this project, 276 samples were analyzed for a genome-scale investigation of promoter methylation, exome sequence, DNA copy number, and messenger and microRNA expression. Frequent mutations were revealed in ARID1A, SOX9, and FAM123B, in addition to the expected APC, TP53, SMAD4, PIK3CA, and KRAS mutations as shown in Figure 1b. Furthermore, amplifications in ERBB2 and the “newly-discovered” IGF2 that might be drug-targeted were also identified in the same project, are two examples of recurrent copy-number alterations.
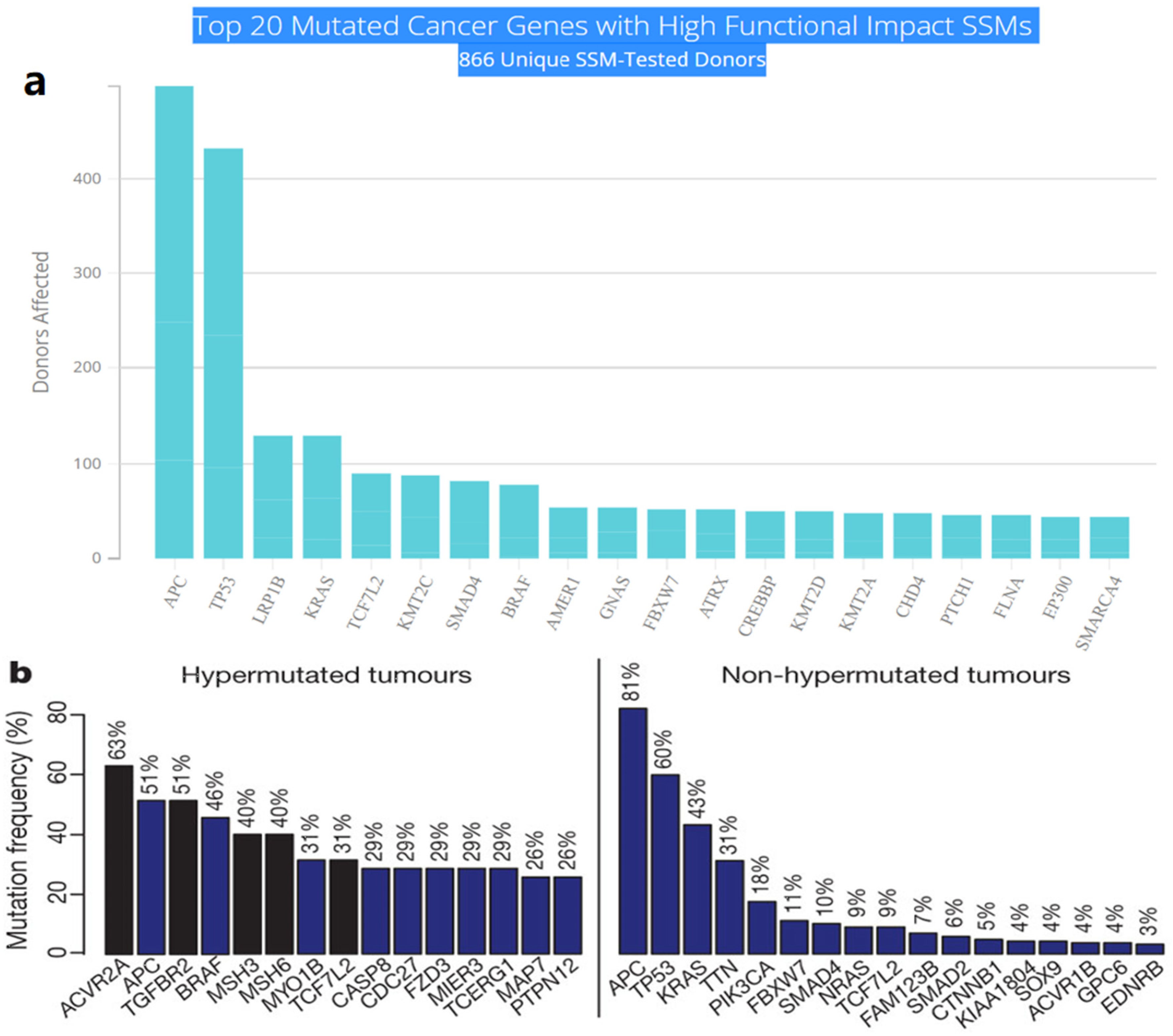
Figure 1. (a) The top 20 mutated genes with high functional impact involved in colorectal cancer extracted from the ICGC Data Portal in three projects: Colon Adenocarcinoma—TCGA, US, Adenocarcinoma, non-Western (China), Rectum Adenocarcinoma—TCGA, US. https://dcc.icgc.org/ (accessed on 15 December 2021) (b) Significantly mutated genes in hypermutated and non-hypermutated tumors adopted from The Cancer Genome Atlas Network [7].
The genome-wide investigations strongly confirm the links between commonly altered driver genes and human colorectal cancer. Tumorigenesis is generated in the presence of mutant driver genes such as APC, KRAS, SMAD4, TP53, PIK3A, ARID1A, and SOX9, in intestinal epithelial cells using organoid culture systems [7][22]. In addition to the previously stated genes, other changed genes identified to be implicated in colorectal cancer carcinogenesis include FBXW7, BRAF, TCF7L2, PIK3CA, GNAS, CBX4, ADAMTS18, TAF1L, CSMD3, ITGB4, LRP1B, and SYNE1 [23]. APC, KRAS, BRAF, PIK3CA, SMAD4, and TP53 are the six CRC driver genes, with APC, KRAS, PIK3CA, and p53 being the most often altered. Mutations in APC, KRAS, and BRAF occur early in the transition phase from normal epithelium to adenoma, whereas PIK3CA mutation and loss of SMAD4 and P53 (due to mutations or epigenetic silencing) occur late, allowing tumor cells to invade surrounding tissues and metastasize, transforming the adenoma into a carcinoma. Mutations in APC, TP53, and KRAS, as well as, to a lesser extent, SMAD4, are related to metastatic conditions while being highly associated with MSI [24]. The APC (adenomatous polyposis coli) gene is thought to be the gatekeeper gene for CRC, with mutations reported in 83% of all cases [25]. KRAS contributes significantly to carcinogenesis by activating the RAF–MAPK and PI3K pathways. TGF-β signaling, on the other hand, promotes epithelial cell differentiation, acting as a tumor suppressor in colorectal cancer. Furthermore, FBXW7 is a component of the ubiquitin ligase complex, which eliminates proto-oncogene products by degradation, acting as a tumor suppressor, and Fbxw7 disruption promotes intestinal carcinogenesis. According to recent findings, mutant p53 affects gene expression globally via a gain-of-function mechanism, which promotes cancer [22]. APC mutations frequently occur concomitantly with KRAS or TP53 mutations, or both. This triad predicts poor prognosis, whereas BRAF, ITGB4, CBX4, CSMD3, SYNE1, FBXW7, and TAF1L are substantially linked to MSI but not to metastatic illness [20].
This entry is adapted from the peer-reviewed paper 10.3390/biom12070878
References
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799.
- Bogaert, J.; Prenen, H. Molecular genetics of colorectal cancer. Ann. Gastroenterol. 2014, 27, 9.
- Issa, J.-P. CpG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993.
- Testa, U.; Castelli, G.; Pelosi, E. Genetic alterations of metastatic colorectal cancer. Biomedicines 2020, 8, 414.
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197.
- Markowitz, S.D.; Bertagnolli, M.M. Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460.
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012.(5). Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 1.
- Lin, M.; Whitmire, S.; Chen, J.; Farrel, A.; Shi, X.; Guo, J.-T. Effects of short indels on protein structure and function in human genomes. Sci. Rep. 2017, 7, 1–9.
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724.
- Garraway, L.A.; Lander, E.S. Lessons from the Cancer Genome. Cell 2013, 153, 17–37.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Loganathan, L.; Muthusamy, K.; Jayaraj, J.M.; Kajamaideen, A.; Balthasar, J.J. In silico insights on tankyrase protein: A potential target for colorectal cancer. J. Biomol. Struct. Dyn. 2018, 37, 3637–3648.
- Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting cancer pathways to tumor engines: A stratification tool for colorectal cancer combining human in vitro tissue models with boolean in silico models. Cancers 2019, 12, 28.
- Kather, J.N.; Poleszczuk, J.; Suarez-Carmona, M.; Krisam, J.; Charoentong, P.; Valous, N.A.; Weis, C.-A.; Tavernar, L.; Leiss, F.; Herpel, E. In silico modeling of immunotherapy and stroma-targeting therapies in human colorectal cancer. Cancer Res. 2017, 77, 6442–6452.
- Greenhalgh, K.; Ramiro-Garcia, J.; Heinken, A.; Ullmann, P.; Bintener, T.; Pacheco, M.P.; Baginska, J.; Shah, P.; Frachet, A.; Halder, R.; et al. Integrated In Vitro and In Silico Modeling Delineates the Molecular Effects of a Synbiotic Regimen on Colorectal-Cancer-Derived Cells. Cell Rep. 2019, 27, 1621–1632.e1629.
- Nazempour, N.; Taleqani, M.H.; Taheri, N.; Najafabadi, A.H.H.A.A.; Shokrollahi, A.; Zamani, A.; Fattahi Dolatabadi, N.; Peymani, M.; Mahdevar, M. The role of cell surface proteins gene expression in diagnosis, prognosis, and drug resistance of colorectal cancer: In silico analysis and validation. Exp. Mol. Pathol. 2021, 123, 104688.
- Fadaka, A.O.; Klein, A.; Pretorius, A. In silico identification of microRNAs as candidate colorectal cancer biomarkers. Tumor Biol. 2019, 41, 1010428319883721.
- Raskov, H.; Søby, J.H.; Troelsen, J.; Bojesen, R.D.; Gögenur, I. Driver gene mutations and epigenetics in colorectal cancer. Ann. Surg. 2020, 271, 75–85.
- Vogelstein, B.; Kinzler, K.W. The path to cancer—Three strikes and you’re out. N. Engl. J. Med. 2015, 373, 1895–1898.
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y. Combined mutation of Apc, Kras, and Tgfbr2 effectively drives metastasis of intestinal cancer. Cancer Res. 2018, 78, 1334–1346.
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.A.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743.
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697.
- Joseph, R.; Little, P.; Hayes, D.N.; Lee, M.S. Characterization of the Number and Site of APC Mutations in Sporadic Colorectal Cancer; American Society of Clinical Oncology: Alexandria, VA, USA, 2017.
This entry is offline, you can click here to edit this entry!