Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biophysics
Pentameric ligand-gated ion channels (pLGICs) mediate or modulate fast synaptic communication in the central and peripheral nervous systems making them vital for neurological processes ranging from memory and learning to nicotine addiction.
- pentameric ligand-gated ion channels
- lipid binding sites
- l
1. Introduction
Pentameric ligand-gated ion channels (pLGICs) mediate or modulate fast synaptic communication in the central and peripheral nervous systems making them vital for neurological processes ranging from memory and learning to nicotine addiction [1,2,3,4]. pLGICs respond to the binding of neurotransmitters by transiently opening either cation- or anion-selective ion channels across the post-synaptic membrane, with prolonged exposure favoring a non-conductive desensitized state(s). The relative stabilities of the resting, open and desensitized states, as well as the rates of inter-conversation between them, shape the magnitude and temporal nature of the agonist-induced response to establish effective inter-neuronal or neuromuscular communication. pLGICs are also targeted by a variety of exogenous molecules that allosterically modulate the agonist-induced response in a manner that alters synaptic communication [5,6].
Lipids are potent activators and/or modulators of ion channels including inward rectifying potassium channels, voltage-gated potassium channels, transient receptor potential channels, mechanosensitive ion channels and pLGICs [7]. The functional sensitivity of pLGICs to membrane lipids was first shown in the 1970s through studies that sought to identify the structural features in the muscle-type Torpedo nicotinic acetylcholine receptor (nAChR) that are responsible for both agonist binding and channel gating. These studies showed that to retain both binding and gating, cholate-solubilized receptors must be purified in the presence of lipids and then placed in a bilayer with a particular lipid composition [8,9]. Since then, the effects of lipids on Torpedo nAChR function have been characterized extensively [10,11,12]. More recently, studies of pLGIC–lipid interactions have extended to other members of the super-family, including the prokaryotic Gleobacter violaceus ligand-gated ion channel, GLIC, and Erwinia chrysanthemi (now Dickeya dadantii) ligand gated ion channel, ELIC.
Over the past 15 years, increasing numbers of structures have shed light on the modes of lipid binding to pLGICs, thus providing a structural context for interpreting functional data on pLGIC–lipid interactions [13].
2. pLGIC Structure
Both eukaryotic and prokaryotic pLGICs exhibit a common architecture consisting of five subunits arranged either symmetrically (homomeric) or pseudo-symmetrically (heteromeric) around a central ion channel pore (Figure 1). In humans, there are four main families of pLGICs that conduct either cations or anions leading to either excitatory or inhibitory post-synaptic responses, respectively. The excitatory cation-selective pLGICs respond to the neurotransmitters, acetylcholine (nicotinic acetylcholine receptor, nAChR) and serotonin (serotonin receptor, 5-HT3R), while the inhibitory anion-selective pLGICs respond to γ-aminobutyric acid (GABA receptor, GABAAR) and glycine (glycine receptor, GlyR). Humans also uniquely express a cation-selective zinc-activated channel, ZAC [14]. Each of the four main families includes a variety of functional hetero- and homo-pentamers that form from different combinations of the sixteen distinct nAChR subunits (α1–α7, α9–α10, β1–β4, δ, γ, and ε), the five distinct 5-HT3AR subunits (A–E), the nineteen distinct GABAAR subunits (α1–α6, β1–β3,γ1–γ3, ε, δ, π, θ, and ρ1–ρ3) or the five distinct GlyR subunits (α1–α4, and β). Each combination leads to a pLGIC with a unique electrophysiological and pharmacological fingerprint. Receptors with different subunit are also targeted to specific cell types and/or regions of the brain [15].
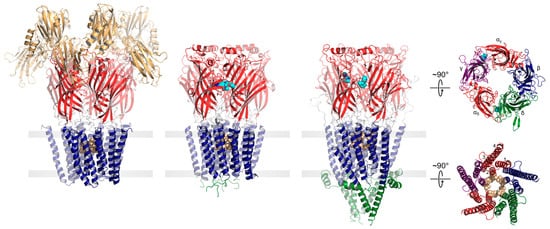
Figure 1. pLGICs display a conserved core architecture with diverse auxiliary features. Side views of the prokaryote DeCLIC (PDB: 6V4S, far left), the human α1β3γ2 GABAAR (PDB: 7QNE, middle left), and the Torpedo nAChR (PDB: 7QL5, middle right) colored according to domains (NTD, orange; ECD, red; TMD, blue; ICD, green). Bound agonists are presented as cyan spheres at the interfaces between two subunits. In the side views, the principal subunit is on the left and the complementary subunit is on the right. Residues forming the channel gate are presented as tan spheres. Top-down views of the Torpedo nAChR ECD (top) and TMD (bottom) are shown on the far right colored according to subunit (α, red; β, blue; γ, purple; δ, green).
The core of each pLGIC structure consists of an N-terminal extracellular domain (ECD), which typically defines agonist binding, and a transmembrane domain (TMD), which contains both the ion-selective pore and the channel gate. In each subunit, the ECD contains ten β-strands (β1–β10) that form two β-sheets folded together into a β-sandwich. The TMD from each subunit is formed from four transmembrane 𝛼-helices (M1 to M4), with M2 lining the channel pore, M1 and M3 shielding M2 from the surrounding lipids, and M4 interacting extensively with the lipid bilayer. Human pLGICs exhibit an intracellular domain (ICD), located between M3 and M4, that interacts with the cytoskeleton. In cation-selective pLGICs, the ICD starts with a short MX 𝛼-helix oriented parallel to the bilayer surface that participates in lipid binding, followed by a mainly disordered region and then a long amphipathic 𝑎-helix, termed MA, which is contiguous with M4 in most cation-selective pLGICs [16]. In anion-selective pLGICs, electron density has not yet been observed for the ICD, making it uncertain whether the MX and MA 𝛼-helices are conserved in these pLGICs [17,18]. Prokaryotic pLGICs lack the ICD, but in some cases contain extra N-terminal domains located just prior to the ligand-binding ECD. Many of these N-terminal domains exhibit sequence similarities to periplasmic binding proteins, possibly allowing these pLGICs to participate in chemotaxis and/or quorum sensing [19].
Agonist sites are typically formed from a series of loops in the ECD extending from the interfaces between the principal and complementary subunits (Figure 1). Agonist binding leads to a compression of these loops around the bound agonist, which drives a conserved rocking motion of the adjacent β-sandwich. The agonist-induced motions of the β-sandwich ultimately translate to the TMD through the covalent link between β10 and M1 and through non-covalent interactions between the β1–β2, β6–β7, and β9–β10 loops and the M2–M3 linker to open the channel gate [20,21,22]. In the resting state, conserved hydrophobic residues in the extracellular half of M2 create an unsolvated energetic barrier that prevents ion flux into the cell [23]. Concerted movements at the ECD–TMD interface lead to a tilting and twisting of the M2 pore lining α-helices away from the central pore axis, thus widening the hydrophobic barrier to allow the diffusion of hydrated ions down their electrochemical gradient [24,25,26,27,28].
Prolonged exposure of pLGICs to agonist leads to the formation of a desensitized state(s) that binds agonist with high affinity, but does not open in response to agonist binding [29]. In anion-selective pLGICs, desensitization arises from a constriction of a gate located near the intracellular end of the transmembrane pore [30]. Loops at the interface between the ECD and TMD and residues near the desensitization gate both govern the rates of pLGIC desensitization [30,31]. Of particular relevance to this review, lipid binding adjacent to M4 influences the rates of desensitization in the prokaryote, ELIC (see below) [32].
3. Nicotinic Acetylcholine Receptors
3.1. Functional Sensitivity of the nAChR to Lipids
Cell-based assays and mutagenesis indirectly suggest that numerous members of the nAChR family exhibit a functional sensitivity to lipids [33,34,35,36,37,38,39,40]. In addition, functional measurements using the Torpedo nAChR reconstituted into liposomes with defined lipid compositions show definitively that a broad range of lipids influence the agonist-induced response, and do so through complex mechanisms. For example, ternary lipid mixtures containing phosphatidylcholine (PC) and both cholesterol and anionic lipids support a robust agonist-induced response, while PC membranes lacking both lipids lock the nAChR in a non-responsive uncoupled conformation that binds agonist but does not normally undergo agonist-induced conformational transitions [41].
The above observation was interpreted to suggest that both cholesterol and anionic lipids are essential for nAChR function and that both lipids exert their functional effects by binding to distinct allosteric sites, a view still prevalent in the literature. Three subsequent observations, however, suggested that neither lipid/lipid-type is essential for the nAChR to undergo agonist-induced conformational transitions. First, increasing levels of either cholesterol or the anionic lipid, phosphatidic acid (PA), in a PC membrane stabilize an increasing proportion of agonist-responsive nAChRs, although PA is more effective in this regard [42,43]. Second, in the presence of anionic lipids a variety of neutral lipids substitute for cholesterol in supporting nAChR function [13,44,45]. Finally, in the presence of cholesterol a variety of anionic lipids substitute for PA in supporting a functional nAChR. Collectively these observations show that if both cholesterol and anionic lipids influence function by binding to distinct allosteric sites, then the lipid specificities for these sites are low and their occupancies not absolutely required for an agonist-induced response.
There are also intriguing differences in the capacities of anionic lipids to influence the agonist-induced response. For example, PC membranes containing high levels of PA stabilize a large proportion of agonist-responsive nAChRs, while PC membranes containing similar levels of phosphatidylserine (PS) or other anionic lipids do not [46,47,48]. These and other observations [43] suggest that PA has a unique capacity to stabilize an agonist responsive nAChR. One possibility is that the small anionic headgroup allows PA to bind with higher affinity and thus greater occupancy to an allosteric site to promote channel function. Another is that high levels of PA increase the ordering of the surrounding bilayer, possibly in a manner that mimics the ordering observed in the presence of cholesterol [48]. High levels (40 mol%) of PA in a PC membrane may be particularly effective at stabilizing a functional nAChR because PA exhibits both the required anionic headgroup charge and an ability to influence bulk membrane physical properties in a manner that supports agonist-induced conformational transitions. Further supporting a role for bulk membrane physical properties in nAChR function, hydrophobically thick PC membranes promote conformational transitions even in the absence of cholesterol and anionic lipids [49].
3.2. Sites of Lipid Action at the nAChR
Early biophysical and computational studies suggested that cholesterol, and possibly other lipids, bind to both annular and non-annular sites on the nAChR to influence function [50,51,52]. Annular sites are those located at the periphery of the TMD in rapid exchange with lipids in the bulk membrane environment, while non-annular sites are those buried between TMD α-helices that are shielded from bulk membrane lipids [53]. In contrast to the plethora of annular lipid sites observed in nAChR structures (discussed below), none of the nAChR structures solved to date exhibits density attributed to buried non-annular lipids. Both the abundance of observed annular lipid sites, which should be more mobile than non-annular lipids, and the absence of observed buried non-annular lipids argue against the existence of functional non-annular lipid binding to the nAChR. On the other hand, structures of the nAChR and other pLGICs reveal density due to annular lipids, but with the acyl chains extending in between TMD 𝛼-helices (see below). In these cases, the distinction between annular and non-annular lipid binding is blurred.
The first direct structural evidence for annular lipid binding to the nAChR was obtained from cryo-electron microscopy (cryo-EM) structures of the detergent-solubilized human neuronal 𝛼4𝛽2 nAChR (both 𝛼43𝛽22 and 𝛼42𝛽23 stoichiometries) and the azolectin nanodisc-reconstituted human neuronal 𝛼3𝛽4 nAChR, the two sets of structures solved in the presence of the water-soluble cholesterol analog, cholesterol hemisuccinate (CHS) [54,55]. Each structure exhibits regions of electron density at the periphery of the TMD that was modeled as cholesterol (Figure 2). The bound cholesterol, located in the cytoplasmic leaflet at both the M4–M1 and the M4–M3 interfaces of each subunit, is close to residues covalently labeled in the Torpedo nAChR by a photoactivatable cholesterol probe [56]. Notably, the electron density attributed to cholesterol disappears when the cryo-EM samples are prepared in the absence of CHS.
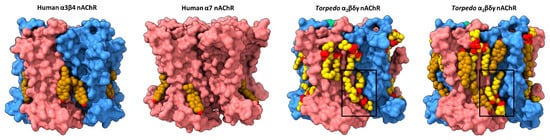
Figure 2. Lipid binding to both extracellular and intracellular leaflet sites on the nAChR. Side views of the TMD for the cholesterol-bound α3β4 nAChR (PDB: 6PV7, far left) and α7 nAChR (PDB: 7EKI, middle left), phospholipid-bound Torpedo nAChR (PDB: 7QL5, middle right), and cholesterol- and phospholipid-bound Torpedo nAChR (PDB: 7SMQ, far right) represented as surfaces, with principal and complementary subunits colored in pink and blue, respectively. Bound cholesterol (brown) and phospholipids (yellow) are presented as spheres with oxygen, nitrogen, and phosphorus colored in red, blue, and orange, respectively.
Annular cholesterol sites are observed in cryo-EM structures of the azolectin nanodisc-reconstituted Torpedo nAChR, which were solved using receptors purified from native Torpedo membranes [57]. Three endogenous cholesterol sites, deemed high affinity, are observed bound to an intracellular leaflet hydrophobic pocket framed by M4, M3 and MX on the principal face of the two 𝛼 subunits and the single 𝛽 subunit. In all three cases, the planar sterol ring is sandwiched between a valine and an arginine on M3 (e.g., 𝛼V294 and 𝛼R301 with 𝛼R301 projecting towards the hydroxyl of cholesterol), and a valine/isoleucine and phenylalanine on MX (e.g., 𝛼V312 and 𝛼F316). Each of the observed cholesterol binding poses overlaps with, but is distinct from, those observed at the M4–M3 interface in the neuronal 𝛼4𝛽2 and 𝛼3𝛽4 nAChRs—the distinct poses could reflect improved modeling due to the higher resolution of the Torpedo structures (2.6 Å for the highest resolution Torpedo structure versus 3.3 and 3.5 Å for the 𝛼3𝛽4 and 𝛼4𝛽2 structures, respectively) or different binding of CHS versus cholesterol. Additional cholesterol sites are observed in structures solved in the presence of exogenously added cholesterol. These extra sites, deemed low affinity, are found in the extracellular leaflet where they frame either side of M4.
It is notable that the cholesterol sites observed in the 𝛼3𝛽4, 𝛼4𝛽2 and Torpedo nAChR structures overlap with regions of low electron density in cryo-EM images recorded from native Torpedo post-synaptic membranes, with the low-density regions attributed to bound cholesterol [58,59,60]. The bound cholesterol is observed at both inner and outer leaflet transmembrane sites. Interestingly, the presence of cholesterol stabilizes a “splayed-apart” arrangement of the M1–M3–M4 α-helices in the outer leaflet of the bilayer, with this arrangement postulated to create space for the pore-lining M2 α-helices to move during gating [59,60]. Cholesterol-interacting regions become more extensive, thus leading to the formation of microdomains in areas bridging adjacent receptors, particularly in the vicinity of the disulfide linkage between δ-δ dimers of neighboring nAChRs.
Annular sites for phospholipids are also observed in each of the Torpedo nAChR structures solved to date, including a conserved inner leaflet site adjacent to, but in some cases overlapping with the high affinity cholesterol sites noted above [57,61,62]. In most cases, the phosphate of the modeled phosphatidylcholine (PC) is sandwiched between two positively charged residues, a conserved arginine located just after the M3 𝛼-helix from the principal subunit and a lysine, arginine, or histidine from the complimentary M4 𝛼-helix (e.g., 𝛼R301 and 𝛾K449 at the 𝛼-𝛾 site; Figure 3). The conserved arginine is positioned by a conserved coordinating tryptophan on the M4 𝛼-helix (e.g., 𝛼W399) and a M3–MX loop histidine (e.g., 𝛼H306), both on the principal face of the lipid binding site. An aromatic side chain from the complimentary M1 𝛼-helix (e.g., 𝛾F242) also forms a stacking interaction with one acyl chain. Note that the binding pose of the choline moiety of the headgroup varies from subunit to subunit and from structures to structure likely because there are no specific coordinating interactions. Furthermore, PC or PA bind quickly to this motif and remain bound for the duration of all trajectories in molecular dynamics (MD) simulations [62]. This site is connected to a salt bridge between M4 and the back of the M2 𝛼-helix, previously shown to be important in channel gating in the human adult muscle nAChR [63]. The five noted residues may constitute a signature motif for a functionally important phospholipid binding site that could have a particularly high affinity for PA.
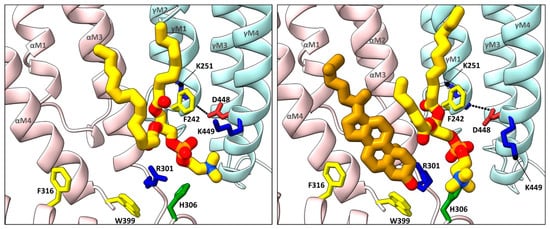
Figure 3. Cholesterol and phospholipids bind to adjacent or overlapping sites in Torpedo nAChR structures. Zoomed in views (defined in Figure 2) of Torpedo nAChR structures with bound phospholipids (PDB: 7QL5) or both phospholipids and cholesterol (PDB: 7SMQ). Subunits and lipids are colored as in Figure 2, with residues interacting with bound lipids represented as sticks colored according to residue type (non-polar, tan; aromatic, yellow; polar, green; cationic, blue; anionic, red). The M2–M4 salt bridge adjacent to the bound lipids is shown as a dashed line.
In the extracellular leaflet, the annular phospholipids typically bind to a shallow cavity formed by a positively charged residue on M3 (e.g., 𝛼K276), the M2–M3 loop and the Cys-loop from the principal subunit along with M1 from the complementary subunit. The bound phospholipid is located between, but in some cases overlaps with the two extracellular leaflet cholesterol sites. It is notable that the outermost M4 𝛼-helix from 𝛼𝛿, and to a lesser extent from 𝛼𝛾, tilts away from the rest of the TMD in agonist bound structures. The outward tilt of M4 allows the acyl chain of an outer leaflet phospholipid to enter the void between the end of M4 and the rest of the TMD where it contacts the strictly conserved Cys-loop FPF motif [62]. The outward tilt of M4 allows the inhibitor d-tubocurarine to bind in the same cavity [57]. It has been proposed that dynamic movements of M4 underlie both the uncoupling of binding and gating that occurs with the Torpedo nAChR reconstituted into PC membranes lacking cholesterol and anionic lipids [46]. Dynamic movements of M4 leading to altered lipid and/or allosteric modulator binding is a common theme observed in several pLGIC structures, as discussed below.
It is intriguing that although the actual number of bound lipids generally increases with the resolution of the solved structure, several bound lipids are observed in each of the nine Torpedo structures solved to date. In contrast, structures of the neuronal 𝛼7 nAChR exhibit either no or only a few bound lipids. Specifically, structures of the 𝛼7 nAChR reconstituted into similar azolectin nanodiscs do not exhibit any bound lipids even though they were solved at relatively high resolutions (2.7 Å to 3.6 Å) [64]. In a second study, diffuse density was observed at the periphery of the TMD of 𝛼7 nAChR structures solved in detergent, with one region of electron density in each subunit modeled as cholesterol (Figure 2) [65]. One speculative interpretation derived from this observation is that the membrane facing surface of the Torpedo nAChR TMD is more amenable to lipid binding than the membrane exposed surface of the 𝛼7 nAChR. An increased propensity to bind lipids could underlie the exquisite functional sensitivity of the Torpedo nAChR to its membrane environment.
Another striking feature of the various Torpedo structures is that, while there is conservation of phospholipid binding sites, which hints at a role for such sites in channel function, the bound cholesterol observed in structures reported by Rahman et al. [57] are not observed in all Torpedo structures, particularly those reported by Zarkadas et al. [62]. The absence of bound cholesterol in the latter structures likely reflects the fact that Zarkadas et al. washed the detergent-solubilized nAChR extensively with detergent-solubilized soybean azolectin during purification leading to cholesterol–phospholipid exchange. The Rahman et al. and Zarkadas et al. Torpedo structures show, not surprisingly, that the protocol used to prepare a pLGIC for cryo-EM imaging can dramatically alter the observed lipid binding. Comparison of the structures also shows that phospholipids and cholesterol bind to overlapping annular sites, thus giving credence to the hypothesis that the low lipid specificity of the Torpedo nAChR results, at least in part, from the binding of different lipids to overlapping sites, albeit with different affinities/occupancies and different efficacies for stabilizing an agonist-responsive nAChR [13,46].
The ensemble of solved nAChR structures also highlight limitations in how structural data can inform our understanding of lipid–nAChR interactions. As noted above, the pattern of bound lipids differs depending on the protocols used to prepare the cryo-EM samples. There may also be subtle differences in lipid binding that result from the different scaffolding proteins used to encapsulate the reconstituted nAChR in a lipid nanodisc. For example, lipid binding in the Zarkadas et al. structures is suggestive of distinct lipid sites, while lipid binding in some of the Rahman et al. structures approaches a continuous annular belt between the M4 𝛼-helices from adjacent subunits (Figure 2). Zarkadas et al. imaged the nAChR embedded in nanodiscs formed using the circular scaffolding protein, MSP2N2, with two molecules of the scaffolding protein surrounding a lipid bilayer with a fixed diameter of 15–17 nm, while Rahman et al. imaged the nAChR in nanodiscs formed using the scaffolding protein, saposin A. The more globular saposin A monomers assemble in different stoichiometries to form nanodiscs with different diameters, depending on the size of the encapsulated membrane protein. It has been suggested that saposin A encapsulates membrane proteins with a minimal number of lipids trapped between the membrane protein and each saposin A monomer [66]—an observation supported by recent structures of the 5-HT3R (see below). The additional lipids observed in structures solved by Rahman et al. could partly reflect a tighter saposin A nanodisc that prevents the diffusion of encapsulated lipids within the nanodisc, thus allowing the detection of both high affinity allosteric and lower affinity annular sites.
Finally, it is significant that the virtually superimposable structures, solved by Zarkadas et al. and Rahman et al., in the presence of the agonist carbamylcholine (Carb) were attributed to different physiological states. Prolonged exposure to Carb should lead to a stable desensitized conformation that binds agonist with high affinity but does not flux cations across the membrane, a finding consistent with functional assays performed on the nanodisc reconstituted nAChR. Backbone restrained MD simulations suggest that the pore of the agonist-bound nAChR is hydrated and likely conductive for cations, a finding inconsistent with a non-conductive desensitized conformation [62]. In unrestrained MD simulations, the pore collapses to a non-conductive conformation like that observed in the resting state. Diffuse density, however, was observed in the pore of the Carb and nicotine bound structures. When this density was modeled as lipids, the conformation of the nAChR was stable in backbone unrestrained MD simulations, with the dynamic lipid blocking hydration of the pore and, thus, cation flux.
The simplest interpretation of the MD simulations is that lipid, possibly from lipid vesicles or empty lipid nanodiscs that are destroyed during sample vitrification, lodges in the open pore in the presence of Carb and traps the nAChR in a transient conformation along the reaction coordinate between an open or pre-open state and the desensitized state. A second speculative interpretation is that the structures represent a true desensitized state, but that the blockage of cation-flux upon desensitization results from the diffusion of lipids from the surrounding bilayer into the pore. Note that lipids have been observed bound to the pores of other ion channels, such as the bacterial mechanosensitive channel, MscS, and have been proposed to play a role in mechanosensitive channel gating [7,67]. Lipid headgroup density has also been observed penetrating into the wide open pore of the prokaryotic pLGIC, DeCLIC [19]. Regardless, the lack of clarity regarding the physiological state of the agonist bound structures highlights the ongoing struggle to definitively assign solved structures to physiologically relevant conformations—particularly for lipid-sensitive ion channels. The limitations in our ability to definitively assign structures to physiological states impacts our ability to elucidate state-dependent lipid–nAChR interactions and, thus, fully understand the mechanisms by which lipids influence nAChR function.
This entry is adapted from the peer-reviewed paper 10.3390/biom12060814
This entry is offline, you can click here to edit this entry!