1. Berberine and Derivatives
Berberine (IUPAC: 16,17-dimethoxy-5,7-dioxa-13-azoniapentacy-clo [11.8.0.0
2,10.0
4,8.0
15,20] henicosa-1(13),2,4(8),9,14,16,18,20-octaene), a benzyl isoquinoline alkaloid (
Figure 5) from the Berberis plant, show a mild antibacterial activity and Fts- Z inhibition. Sun et al. in their work, studied the active site in
S. aureus for Fts-Z (PDB ID—4DXD) binding through in-silico technique and the found interdomain region participating actively in the docking comparable to that of PC190723 [
61]. The planar structure of the compound is best suited for interaction with the C-terminus beta sheet of Fts-Z protein. Further they designed and synthesized 9-phenoxyalkyl substituted derivatives of berberine (
Figure 6) and found a compound that has a high potent activity which was further verified through in-vitro antimicrobial susceptibility assay and GTPase activity assay. Dasgupta and colleagues, a few years earlier had reported the inhibitory activity of berberine through In-vitro isothermal colorimetry (ITC) and STD NMR spectroscopy [
75]. The studies indicated the overlap of the binding site of berberine with the GTP binding pocket in the Fts-Z protein. Berberine also had low toxicity rates and some other side effects like that of bilirubin -induced brain damage causing jaundice in infants and expecting mothers [
87,
96,
97].
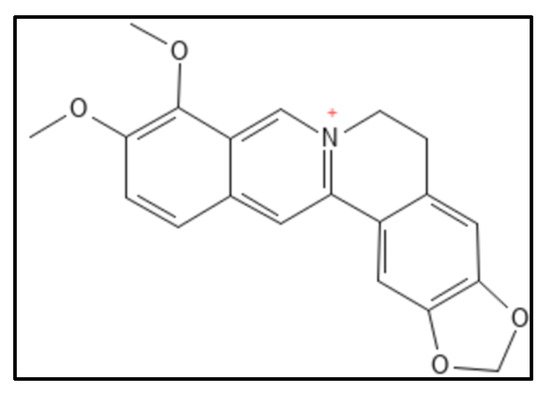
Figure 5. Berberine structure; Mol. Formula—C
20H
18NO
4+; Mol. Wt.—336.4; Experimental details—berberine-dependent Fts-Z assembly observed (IC
50 = 10.0 ± 2.5 µM); Fts-Z GTPase activity observed (IC
50 = 16.01 ± 5.0 µM), ITC results—dissociation constant (K
D = 0.023) entropy driven [
61]. Source: PubChem-CID 2353.
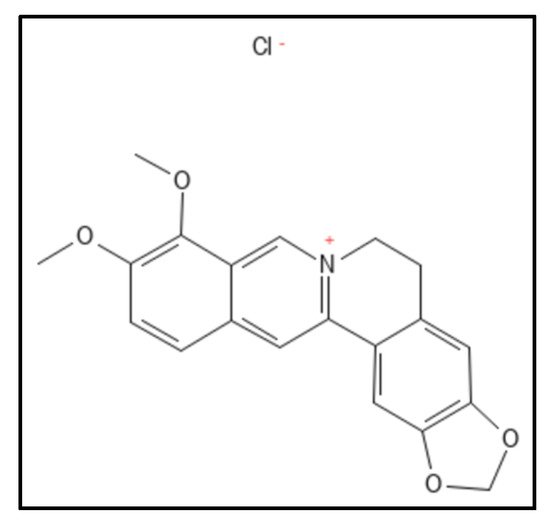
Figure 6. Berberine chloride derivative; Mol. Formula—C
20H
18ClNO
4; Mol. Wt.—371.8; Experimental details—Inhibited MRSA ATCC 29247 (MIC = 2 µg mL
−1), ATCC 700221 (MIC = 4 µg mL
−1),
E. coli (MIC = 32 µg mL
−1),
K. pneumoniae (MIC = 64 µg mL
−1); showed GTPase (IC
50 between 37.8 and 63.7 µM). In-silico studies done on PDB-4DXD (interacting residues were Ile197, Leu200, Val203, Leu209, Met226, Leu261, Val297 and Ile311) [
61]. Source: PubChem-CID 12456.
2. Sanguinarine
Sanguinarine (IUPAC: 24-methyl-5,7,18,20-tetraoxa-24-azoniahexacy-clo [11.11.0.0
2,10.0
4,8.0
14,22.0
17,21] tetracosa-1(24),2,4(8),9,11,13,15,17(21),22-nonaene) is a benzophenanthridine alkaloid from rhizome of
Sanguinaria canadensis is found to inhibit cytokinesis in bacteria. It inhibits the
E. coli Fts-Z assembly by perturbing the Z ring and was also found to inhibit
B. subtilis cell growth without affecting nucleoid segregation at an IC
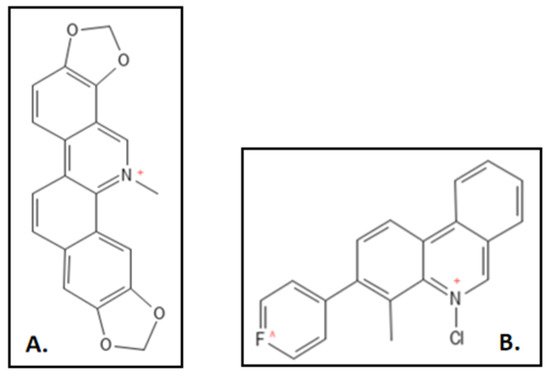
50 of 3µM. This interaction was investigated through in-vitro size exclusion chromatography and fluoroscent probing. Mutational studies were also conducted by the group (
Figure 7A) [
90]. A representative compound 21 had 2 fold higher GTPase activity as compared to sanguinarine for which a patent was also filed [
98]. Liu and coworkers designed and synthesized sequence of novel 5-methyl-2-phenylphenanthridium (
Figure 7B) derivatives by simplifying the skeleton of sanguinarine. These had an exceptionally elevated anti-bacterial activity against an array of 10 antibiotic sensitive and resistant strains. MIC values for
S. aureus ATCC25923,
S. epidermis,
S. pyogenes PS and
PR,
B. subtilis ATCC9372,
B. pumilus ATCC63202 were ranged from 0.06 to 8 and 0.25–16 µg mL
−1 respectively and for
E. coli ATCC 25922 and
P. aeruginosa ATCC27853 it was more than 64 µg mL
−1 [
89].
Figure 7. (
A) Sanguinarine; Mol. Formula—C
20H
14NO
4+; Mol. Wt.—332.3; Experimental details—Dissociation constant (K
D) = 18–20 µM; IC
50 = 3 ± 1 µM for
B. subtilis 168, 14 ± 2.3 µM for
E. coli BL21 [
89]. (
B) 5-methyl-2-phenylphenanthridium derivative of Sanguinarine [
97]. Source: PubChem-CID 5154; PMID-29657101.
3. Cinnamaldehyde and Derivatives
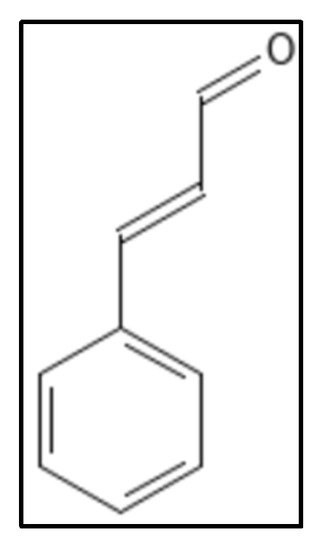
Cinnamaldehyde (IUPAC: (E)-3-phenylprop-2-enal), (
Figure 8) a plant derived product from spices (stem bark of
Cinnamomum cassia) has shown an array of potential medicinal properties. It has been reported to possess inhibitory activity against yeasts, filamentous molds and many bacteria through various pathways including inhibition of cell wall biosynthesis, changing of membrane structure and integrity and inhibiting GTPase activity [
63]. Domadia and coworkers reported in-vitro, in-silico and in-vivo activity of cinnamaldehyde to perturb Z ring morphology and GTP hydrolysis with an affinity of 1.0 ± 0.2 µ/M [
76]. Molecular modeling results were in concordance with STD-NMR results showing binding at the T7 loop in the C-terminal region. The results of MSA (multiple sequence alignment) also showed some of the residues- R202, V208, N263, G295, N263 to be highly conserved among Fts-Z. Xin and coworkers worked on derivatives of cinnamaldehyde where they found that substitution in the benzene ring at ortho or para position of cinnamaldehyde, by small groups increased the activity of compounds so synthesized against different microbes. They further stated that 2-methyl benzimidazoyl moiety was had a better efficiency against all strains, they tested and gave three compounds (marked as 3, 8, 10 in [
94]) which had MIC of 4 µg mL
−1 in two compounds and 10 µg mL
−1 against
S. aureus (ATCC 25923). Two compounds (marked as 4, 10 in [
64]) gave MIC (4 µg mL
−1)values 32 times better than the reference drugs used.
Figure 8. Cinnamaldehyde structure. Mol. Formula—C
9H
8O; Mol. Wt.—132.16; Experimental details—inhibits
E. coli (MIC = 1000 mgL
−1),
B. subtilis (MIC = 500 mgL
−1), MSRA (MIC = 250 mgL
−1), GTPase inhibition (IC
50 = 5.81 ± 2.2 µM). In-silico docking (PDB ID—1FSZ), interacting residue were V208, R202, N263, G295, S297 [
89]. Source PubChem: CID 637511.
In 2015, another group of researchers took this study one step further by synthesizing cinnamaldehyde derivatives and reported comparable activity of them wherein some possessed cell division inhibition properties in
S. aureus between 0.25–4 µg mL
−1. They found good activity when 2-methylbenzimidazolyl substitution was at the first position and 2,4-dichlorophenyl was at the third position. Polymerization inhibition and GTPase activity of
S. aureus Fts-Z were shown in dose-dependent manner through biological assays of compounds [
94].
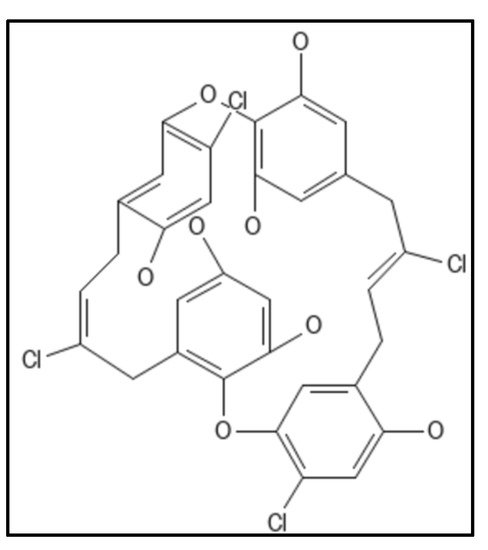
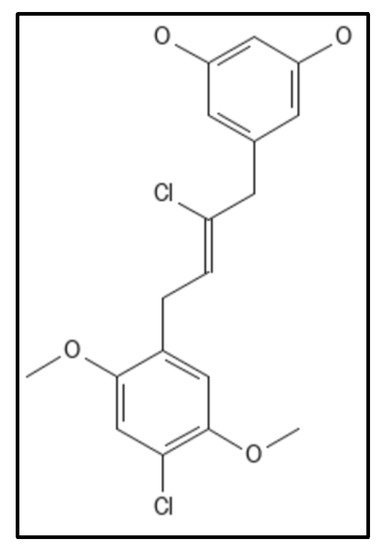
4. Chrysophaentins
Chrysophaentins are group of anti-infectives obtained from marine sources, chrysophyte alga,
Chryosphaeum taylori, including 8 phytochemicals (A-H), showed the inhibitory activity for MRSA, VREF (vancomycin-resistant
E. faecium) and
S. aureus [
93]. Of these, Chrysophaentin A (IUPAC: (9E,25E)-4,10,20,26-tetrachloro-2,18-dioxapentacy-clo [2 6.2.2.13,7.119,23.012,17]tetratriaconta-1(30),3,5,7(34),9,12(17),13,15,19,21,23(33),25,28,31-tetradecaene-6,14,16,22,30,31-hexol), (
Figure 9) showed the most potent antibacterial activity and also inhibited GTPase activity in
E. coli which was attributed to the presence of hydroxyl group. Also, Chrysophaentins A had 12-fold higher MIC
50 as compared to Chrysophaentins D, due to the presence of chlorine at chains A and C. Another compound hemi-Chrysophaentins(
Figure 10) was synthesized and reported to have a reaction system of Chrysophaentins A [
67].
Figure 9. Chrysophaentin A Structure. Mol. Formula—C
32H
24Cl
4O
8; Mol. Wt.—678.3; Experimental details—GTPase activity (IC
50 = 6.7 ± 1.7 µg mL
−1); MIC
50 = 1.8 ± 0.6 µg mL
−1, 1.5 ± 0.7 µg mL
−1, and 1.3 ± 0.4 µg mL
−1 against SA, MRSA, and multi drug resistant SA (MDR-SA), respectively; and 3.8 ± 1.9 µg mL
−1 and 2.9 ± 0.8 µg mL
−1 toward
E. faecium and VREF; In-silico docking on homology modelled E.coli, placed compound in GTP binding site (residues Gly20, Asn24, Asn 43, Ala 70, Thr 108, Arg 142 formed H-bond) [
93]. Source: PubChem-CID 46872004.
Figure 10. Hemichrysophaentin Structure; Mol. Formula—C
18H
18Cl
2O
4; Mol. Wt.—369.2 [
93]. Source: PubChem-60164930.
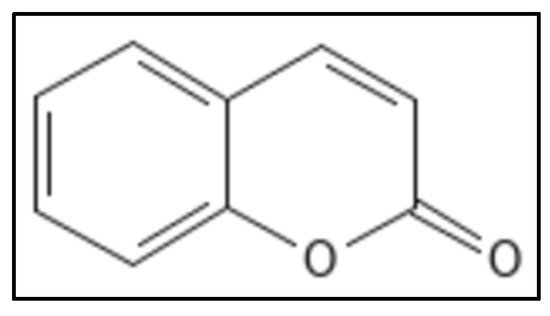
5. Coumarins
Coumarins (IUPAC: chromen-2-one), (
Figure 11) are 1,2-benzopyrone derivatives were derived from different plants with a proven Fts-Z inhibition activity. They have a 2H-chromen-2-one composed of lactone and aromatic ring, along with 2 oxygen atoms forming bonds with enzyme residues and thus are responsible for pharmacological properties. The aromatic ring also plays role in destabilizing enzymes by forming hydrophobic bonds [
69]. Ammoresinol, anthogenol, ostruthin, novobiocin, chartreusin and coumermycin are some of the coumarins which also show anti-bacterial activity against many Gram-negative and Gram-positive bacteria [
99]. A study suggested that coumarins had potent activity against
M. tuberculosis (H37Rv) by inhibiting GTPase activity and polymerization of Fts-Z. Other coumarins namely, Scopoletin (having IC
50 of 41 μM for Fts-Z polymerization and 23 μM for GTPase activity) and Daphnetin (IC
50 of 73 μM for Fts-Z polymerization and 57 μM for GTPase activity) have proven activity. In-silico studies performed on coumarins and Fts-Z indicate that it binds in the T7 loop of Fts-Z [
100,
101,
102,
103,
104,
105,
106].
Figure 11. Coumarin Structure. Mol. Formula—C
9H
6O
2; Mol. Wt.—146.14; GTPase activity (IC
50 = 212 ± 4.12 µM); Fts-Z polymerization (IC
50 = 200 ± 4.2 µM) [
58]. Source: PubChem-323.
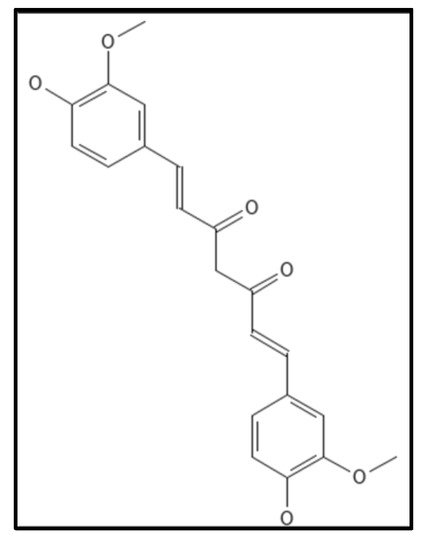
4.6. Curcumin
Curcumin (IUPAC: 1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione/diferuloyl methane), (
Figure 12) is a chromophore in rhizomes of the plant
Curcuma longa (turmeric). It is a polyphenolic compound, possessing a wide spectrum of biological activities and is traditionally used in as a household remedy for curing various diseases, as spice and as a food preservative in South East Asian countries including India [
88]. The presence of two ortho methoxylated phenols having conjugate linking with beta-di-ketone function makes it an appealing target for the drug industry. Rai and coworkers, revealed the inhibitory action of curcumin to polymerize Fts-Z in
B. subtilis wherein Z-ring formation was perturbed and GTPase activity was enhanced [
107]. Roy and colleagues found putative curcumin binding sites in
E. coli Fts-Z and
B. subtilis Fts-Z forming bonds in the GTP binding pocket through the computational docking technique [
108]. Some of the research groups are working on the nano-formulation of curcumin to increase its stability in in-vitro and in-vivo setups. The major drawback of curcumin is its poor aqueous solubility and bioavailability [
92].
Figure 12. Curcumin Structure; Mol. Formula—C
21H
20O
6; Mol. Wt.—368.4; Experimental details—inhibited
B. subtilis 168 (IC
50 = 17 ± 3 µM),
E. coli K12 MG1655 (IC
50 = 58 ± 5 µM); dissociation constant (K
D) = 7.3 ± 1.8 µM [
108]. Source: PubChem-CID 969516.
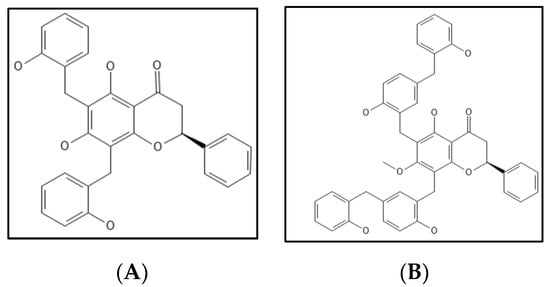
7. Dichamanetin and Derivatives
Dichamanetin (
Figure 13A) is a natural polyphenolic compound obtained/isolated from Uvariachamae and 2‴-hydroxy-5″-benxzylisouvarinol-B (
Figure 13B) is another natural polyphenolic compound from
Xylopia afticana found by two independent researchers [
109]. Dichamanetin and derivatives are structurally similar to Zantrins Z1, and have anti-bacterial activity against many Gram-positive microbes (
S. aureus,
B. subtilis,
M. smegmatis and
E. coli) comparable to Zantrin. Urgaonkar and colleagues examined the impact of dichamanetin on
E. coli Fts-Z GTPase activity and found that the inhibition IC
50 values were 12.5 µM and 8.3 µM, respectively [
109].
Figure 13. (
A) Dichamanetin Structure; Mol. Formula—C
29H
24O
6; Mol. Wt.—468.5; Experimental details—GTPase inhibition in
E.coli (IC
50 = 12.5 ± 0.5 µM) [
109]. (
B) 2‴-hydroxy-5″-benxzylisouvarinol-B [
109]. Source: PubChem-CID 181193.
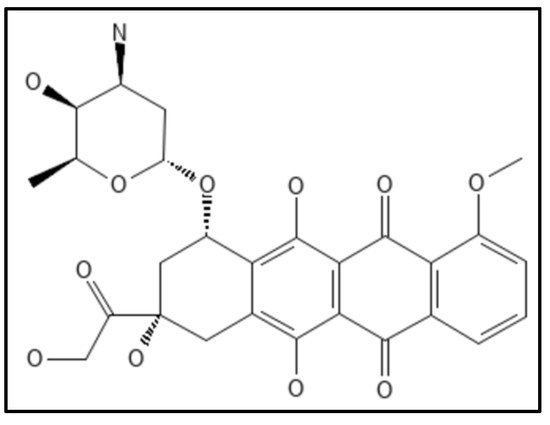
8. Doxorubicin
Doxorubicin, (IUPAC: (7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione) (
Figure 14) an anthracycline antibiotic derived from the actinobacterium
Streptomyces peucetius, has been found as a powerful Fts-Z inhibitor that inhibits
E. coli growth by disrupting Fts-Z functions [
110]. It was identified as a small molecule targeting Fts-Z and suppressing bacterial division utilizing an independent computational, biochemical, and microbiological method from a drug library authorized by the US FDA. The fluorescence-binding experiment indicated that it interacts significantly with Fts-Z without changing membrane structure or nucleoid segregation in microbes. The effects were visible on both GTPase activity and Fts-Z assembly.
Figure 14. Structure of Doxorubicin; Mol. Formula—C
27H
29NO
11; Mol. Wt.—543.5; Experimental details—Antibacterial activity against
E. coli BL21,
B. subtilis (MIC—20 µM, 10 µM respectively); Dissociation constant (
KD 120 ± 61 nM) [
110]. Source: PubChem-CID 31703.
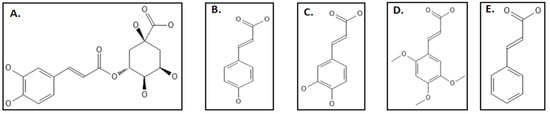
9. Phenylpropanoids
Phenylpropanoids are phytochemicals produced primarily due to stress including wounding, UV irradiation, pollutants, infections, and several other environmental factors to protect them against pathogens and predators. This defensive characteristic can be attributed to their free radical hunting and antioxidant properties [
111]. Studies clearly indicate the use of these phytochemicals for the production of anti-bacterial agents [
111]. Hemaiswarya and coworkers studied the effect of 8 phenylpropanoids (chlorogenic acid, caffeic acid, 2,4,5-trimethoxycinnamic acid, cinnamic acid and p-coumaric acid) (
Figure 15A–E), against
E. coli Fts-Z through both in-silico and in-vitro techniques [
111]. Among them chlorogenic acid which is an ester of quinic aid and caffeic acid was found to have the highest IC
50 value of 69.55 ± 3.6 µM, caffeic acid, cinnamic acid, p-coumaric acid followed with 105.96 ± 6.3 µM, 238.91 ± 7.1 µM, 189.53 ± 3.7 µM, respectively. 2,4,5-trimethoxy cinnamic acid, 3,4-dimethoxy cinnamic acid and eugenol were the least potent (IC
50 < 250 µM.). Light scattering experiments and circular dichroism studies supported the results. The in-silico studies indicated that the binding of phenylpropanoids happens not less than a residue in the T7 loop which is considered to be most important in the Fts-Z structure. Some other compounds which have inhibitory effects against Fts-Z were phenyl acrylamide [
111,
112], vanillin derivative 3a and 4u which have been tested against
S. aureus, S. pyogenes and
M. tuberculosis [
112,
113,
114]
Figure 15. Phenylpropanoids group inhibiting Fts-Z: (A) Chlorogenic acid; (B) p-Coumaric acid; (C) Caffeic acid; (D) 2,4,5-Trimethoxycinnamic acid.; (E) Cinnamic acid. Source: PubChem.
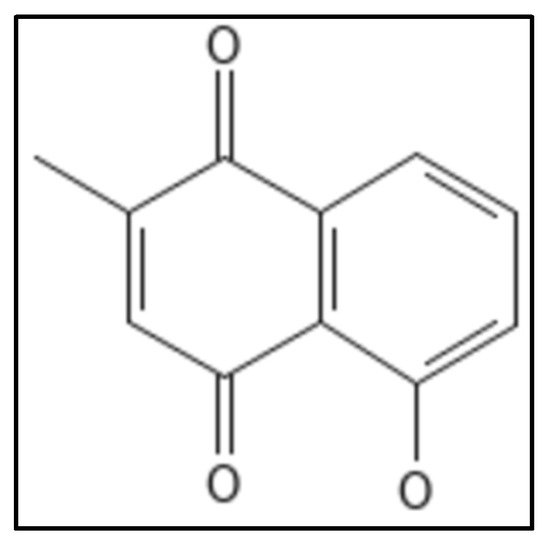
10. Plumbagin
Plumbagin (IUPAC: 5-hydroxy-2-methylnaphthalene-1,4-dione), (
Figure 16) is a phytochemical from the roots of the
Plumbago zeylanica plant [
90]. It is a secondary naphthoquinone derivative known to exhibit a variety of biological activities such as cell proliferation in mammals [
115], fungus [
116] and bacterial cells [
117]. The antimicrobial properties do not affect
E.coli,
S. typhimurium [
118]. Acharya and coworkers, through their ex-vivo experiment demonstrated the binding of plumbagin with cellular microtubules in the colchicine cavity, cell viability experiments gave the IC
50 value of 14.6 µm [
119]. The structural similarity of Fts-Z with that of tubulin has caused many scientists to work on this and plumbagin was found to inhibit Fts-Z in
B. subtilis 168 in a dose dependent manner. Further in-silico studies showed the possible binding site located at residues D199 and V307 [
120].
Figure 16. Plumbagin Structure. Mol. Formula—C
11H
8O
3; Mol. Wt.—188.18; Dissociation constant (K
D) 20.7 ± 5.6 µM [
115]. Source: PubChem-10205.
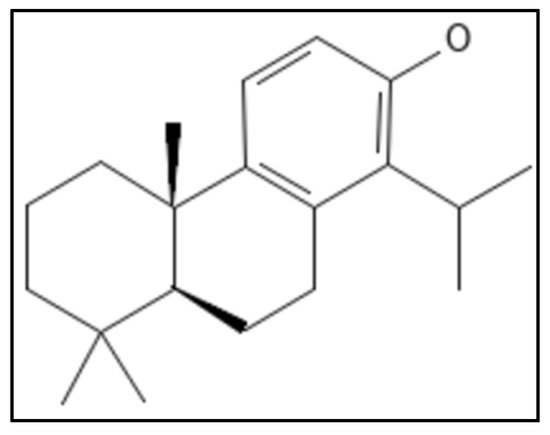
11. Totarol
Totarol, a diterpene phenolic phytocompound, (IUPAC: (4bS,8aS)-4b,8,8-trimethyl-1-propan-2-yl-5,6,7,8a,9,10-hexahydrophenanthren-2-ol) (
Figure 17), is obtained from a conifer (
Podocarpus totara) and showed activity against
M. tuberculosis and
B. subtilis [
121,
122]. Although totarol was shown to have high anti-bacterial action against a variety of Gram-positive bacteria, it had no effect on Gram-negative bacteria and no antifungal properties as well. The experiments done indicated that totarol did not disrupt the
B. subtilis membrane or nucleoid segregation, it only affected the functioning of the Z-ring.
Figure 17. Totarol structure, Mol. Formula—C
20H
30O; Mol. Wt.—286.5; Experimental Details—MIC against
B. subtilis = 2 µM,
S. aureus = 5.4 µM,
M. tuberculosis = 16 µg mL
−1; Dissociation constant K
D = 11 ± 2.3 µM [
121]. Source: PubChem-CID 92783.
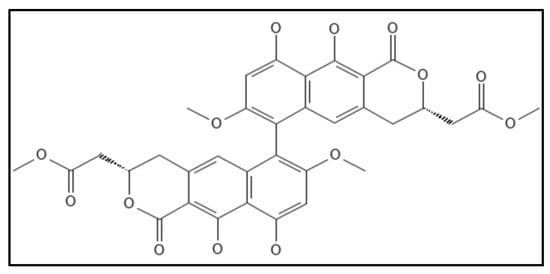
12. Viriditoxin
Viriditoxin (IUPAC: methyl 2-[(3S)-6-[(3S)-9,10-dihydroxy-7-methoxy-3-(2-methoxy-2-oxoethyl)-1-oxo-3,4-dihydrobenzo[g]isochromen-6-yl]-9,10-dihydroxy-7-methoxy-1-oxo-3,4-dihydrobenzo[g]isochromen-3-yl]acetate), (
Figure 18) a cytotoxic compound obtained from fungus
Aspergillus sp. (MF6890) was found to inhibit the bacterial Fts-Z polymerization by Wang and co-workers in 2003 [
14]. The IC
50 values for
E. coli GTPase activity were 7.0 µg mL
−1, for polymerization were 8.2 µg mL
−1 and found to elongate
B. subtilis cells. Another group of researchers isolated this compound from the fungus
Paecilomyces variotii derived from jellyfish and found that viriditoxin stabilized microtubule polymers in SK-OV-3 cells and exhibited antimitotic and antimetastatic potential.
Figure 18. Viriditoxin Structure. Mol. Formula—C
34H
30O
14; Mol. Wt.—662.6; inhibited Fts-Z polymerization (IC
50 = 8.2 μgmL
−1), GTPase inhibition (IC
50 = 7.0 μgmL
−1) [
14]. Source: PubChem-53343291.
Viriditoxin is also effective against a wide range of drug-resistant Gram-positive infections, including
S. aureus (MIC 4–8 µg mL
−1), and
E. faecium (MIC 2–16 µg mL
−1). Inducing Fts-Z expression in bacterial cells may increase the MIC value, suggesting that viriditoxin targets Fts-Z in these bacterial strains. Viriditoxin can induce
B. subtilis cells to elongate, according to morphological research [
94].
This entry is adapted from the peer-reviewed paper 10.3390/biology11050624