Central obesity refers to excessive abdominal fat that builds up around visceral organs and negatively impacts health. It is the key element of the MetS, the common pathological feature in diabetes and cardiovascular disease. Central obesity has more important health consequences than the body mass index in general and the CTS in particular. Apparently, central androgenic obesity does not fit the female predominance of the CTS. This gender distribution was initially explained by the smaller cross-sectional area of the carpal tunnel in women and the fluid retention caused by estrogens.
- obesity
- carpal tunnel syndrome
- metabolic syndrome
- central obesity
- myosteatosis
1. Introduction
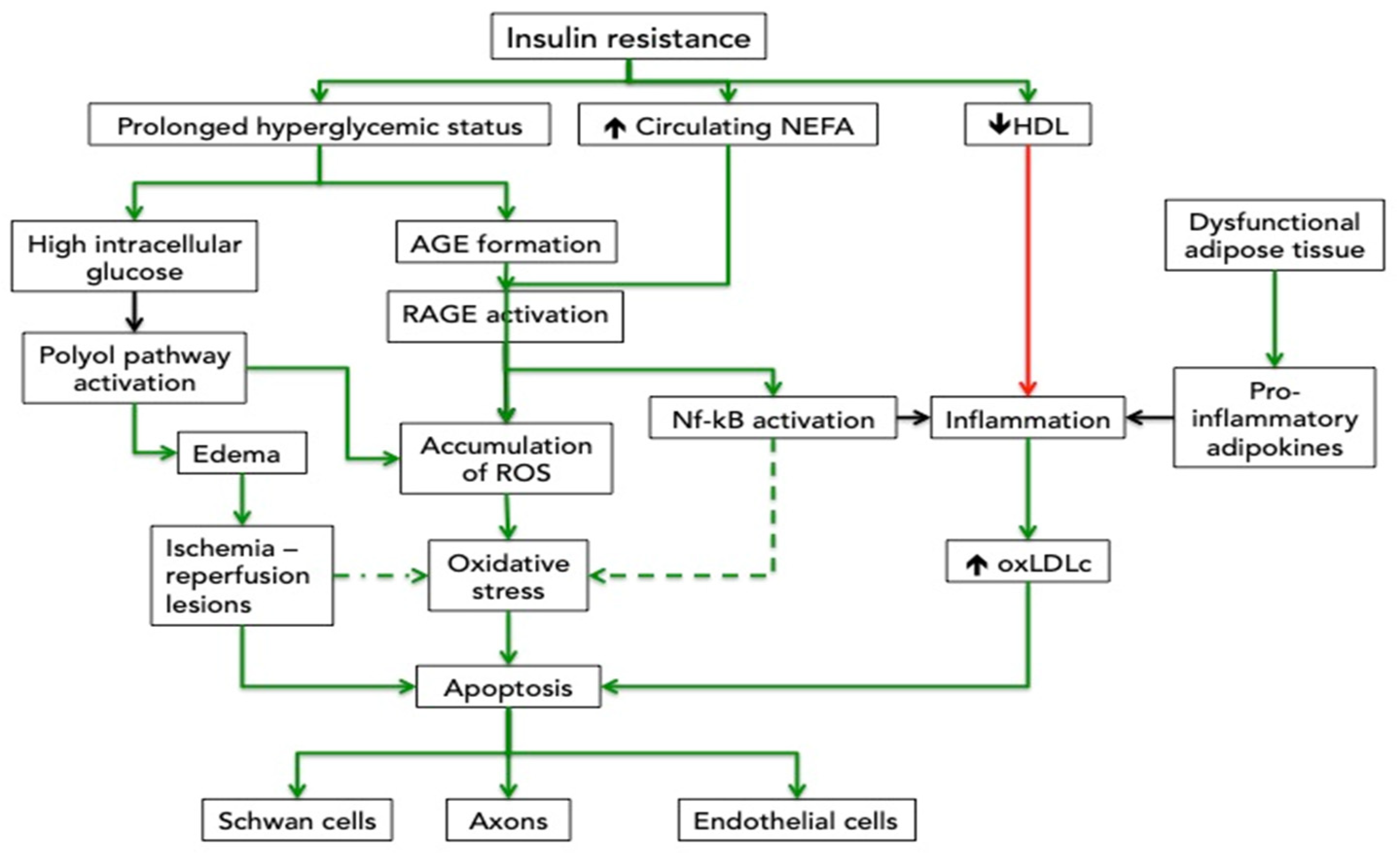
2. Effects of Hyperglycemia on Nerve Impairment
The presence of the MetS almost doubles the chance of peripheral neuropathy [1]. In this study, the relation between MetS and neuropathy was independent of the presence of diabetes but closely related to the waist circumference and triglyceridemia. Thus, searching for and correcting of the MetS components, was proposed for any idiopathic neuropathy [2].
Some researchers found that peripheral neuropathy was only associated with insulin resistance being independent of the MetS [3]. An indirect proof of a long subclinical evolution of neuropathy is the finding of a reduced sensory nerve action potential amplitude in the median nerve in 70% of patients on the first diagnosis of type 2 diabetes mellitus (T2DM) [4].
The neuropathy induced by hyperglycemia combines axonopathy with Schwannopathy features. The decrease in the Na+-K+ pump activity alters the axonal function [5]. The reduced trans-axonal ionic gradient and ionic currents influence neuronal transmission and conduction velocity. The Schwann cells’ dysfunction is responsible for the morphological changes in the myelin sheath, the disruption of the neural support, and the impaired repair of damaged nerves [6].
Several mechanisms explain these modifications. The high intracellular glucose activates other catabolic processes than glycolysis, such as the polyol pathway. The polyol pathway starts with the transformation of glucose into sorbitol, catalyzed by aldolase reductase. This reaction utilizes a hydrogen group donated by NADPH. Sorbitol is then converted into fructose via sorbitol dehydrogenase and donates a hydrogen group to NAD+, maintaining the redox balance. The second reaction does not occur in cells missing sorbitol dehydrogenase, such as the Schwann cells [7]. As a consequence, these cells will acquire an unbalanced NADPH/NAD equilibrium and accumulate reactive oxygen species (ROS). These supplementary ROS are added to the already existing high level of ROS produced by the mitochondria exposed to high levels of intracellular glucose and as well as by the activation of the RAGE inside the neurons. The first organelles damaged by ROS are the mithocondria. Therefore, the axons, which have a large mitochondrial pool and depend on the local energy production, are the neuron parts mostly susceptible to the ROS effects [8]. Besides the oxidative stress, sorbitol is an osmotic compound that attracts water into the cell and produces cellular edema. It has been shown that the activation of the polyol pathway induces oxidative stress, edema, and de-differentiate de Schwann cells, a process which is reversed by an inhibitor of the sorbitol formation [9].
3. Effects of Dyslipidemia on Nerve Impairment
There is also a significant contribution of dyslipidemia to peripheral nerves deterioration, even if the clinical association is less consistent. Some authors found an association between hypertriglyceridemia and subclinical motor and sensory axonopathy, expressed by the delay in distal latencies and decrease in conduction velocities [10][11]. Others have found this association only in those with uncontrolled glycemia or did not find any relation with triglycerides (TG) [12][13]. A systematic review concluded that the association is, however, valid in the subgroup of T2DM [14].
These apparently conflicting results might reflect the independent analysis of the one-by-one components (e.g. TG) of the complex lipid transport system in disregard of the dynamic exchange in lipid content between the lipoprotein particles and the direct and reverse cholesterol movement. TG is an important marker of how much cholesterol is delivered to the peripheral cells but should be considered in relation to the activity of cholesteryl-ester transfer protein, which transfers TG and cholesterol esters to LDL particles, or with the activity of the lipoprotein lipase, which transforms the TG rich LDL particles in small-LDL particles [15]. The reverse transport should be taken into account as well, particularly for the low HDL-c in MetS. Experimental data show that reduction of TG and of the non-esterified fatty acid (NEFA) without normalization of glycemia are able to ameliorate the peripheral neuropathy [16]. Even more, the high level of circulation NEFA increase the superoxide production in human Schwann and endothelial cells [17]. The glycated albumin, which is more abundant in insulin resistance states, facilitates the NEFA penetration of the blood-nerve barrier [18].
The HDL molecule, which is generally in lower concentrations in plasma from patients with MetS, is not only the carrier for reverse cholesterol transport, but has also anti-inflammatory and antioxidant properties [19]. In fact, systemic low-grade inflammation is a characteristic of the MetS and is mainly part of the abnormal secretory pattern of the “unhealthy” adipocytes.
In the inflammatory context of the MetS, the small low-density lipoprotein cholesterol (LDL-C) is more frequently oxidized [20]. Besides the well-known effects on endothelium, the oxidized LDL (oxLDL) could also link to the oxLDL receptor in neurons. During periods of high-fat diet-induced dyslipidemia, the oxLDL receptor in the dorsal root ganglia neurons of mice is activated and contributes to oxidative stress. Even more, in this experiment, the oxLDL effect was additive to the one derived from the hyperglycemic status [21]. The oxidative milieu favors the formation of oxysterols from cholesterol. The oxysterols induce the caspase 8 pathway and apoptosis of the neurons [22].
Peripheral neurons also express the liver X receptors (LXR α/β) (NR1H3 and NR1H2), a cholesterol sensor that works as a transcription factor to control the lipid metabolism [23]. Gavini et al showed that the LXR reduces the proliferation of the Schwan cells in obesity. The effect was directly related to the downregulation of neurogenic 1 by saturated fatty acids [24]. The same group found also that activation of LXR can reduce pain in diet-induced obesity. In the presence of a powerful agonist of LXR, the lipid content of the macrophages from peripheral nerves was reduced with a consequent decline in inflammation and the slower progression of the nerve lesions [25]. All the above, suggest that LXR could explain symptoms of obesity associated CTS, and should be investigated for their therapeutically potential.
Confounder in the relation between dyslipidemia and CTS that must be taken into account in epidemiological studies is hypothyroidism, as on one side, CTS is more frequent in patients with this medical condition and on the other side dyslipdemia, hyperinsulinemia and large waist circumference are present even in mild hypothyroidism [26]. In some studies, the body mass index (BMI) was the link between hypothyroidism and CTS [27][28]. Even in euthyroidism, the TSH level in the upper range significantly increases the chances of MetS [29].
The thyroid hormones are essential for the lipid metabolism. In hypothyroidism, there is an imbalance between cholesterol synthesis and clearance. There are at least thre mechanisms by which the clearance of the LDL is reduced: the diminished synthesis of the LDL-receptor, decreased hepatic cholesterol uptake, and biliary excretion [30]. On the other side, the oxidation of the LDL-receptor increases favors inflammation and oxidative stress [24]. The lipoprotein lipase activity is also impaired in hypothyroidism, because it reduces the TG clearance and increases the circulating VLDL particles [31]. The synthesis of the endogenous cholesterol is not increased while the amount of available cholesterol in the liver is maintained or even made higher because the cholesterol absorption increases and the beta-oxidation in hepatocytes decreases; the overall effect is a higher VLDL output from the liver [32]. TSH increases the activity of the hormone-sensitive lipase and the adipocyte lipolysis, mobilizing more fatty acids in the circulation [33]. The HDL synthesis and maturation are also affected, as the thyroid hormones favor the synthesis of apoA1 and the efflux of cholesterol from macrophages via the ATP-binding cassette A1 [34]. Overall, hypothyroidism increases TG, LDL, and VLDL and reduces the reverse transport and the excretion of cholesterol [35]. To the general effects of dislipidemia on the peripheral nerves, hypothyroidism adds the deposition of pseudo mucinous substances on the median nerve sheath and the decrease in the cross-sectional area of the carpal tunnel [28][36]. The latest effect was reversed by thyroxin treatment.
Fat is also a deposit for neurotoxic occupational hazards and accumulation of these toxicants might delay their excretion. One of the best characterized peripheral neurotoxic with adipose tissue tropism is n-hexane [37]. The chronic n-hexane exposure initiates the formation of lysine adducts with cytoskeletal proteins; the changes in the protein structure interfere with the insertion of newly synthesized neurofilaments into the axonal cytoskeleton and the microtubule-binding and leads to the atrophy of the nerve, sensory and motor impairment [38][39].
Taken together, these general neural dysfunction mechanisms could be included in the expanded understanding of the double crash syndrome, defined as the coexistence of a local compression with a systemic cause of neuropathy [40].
4. Vascular effects
The median nerve is affected by ischemia, secondary to the vascular remodeling or through compression of the nerve inside an abnormal narrow tunnel. Both mechanisms might be the consequences of vascular modifications which characterize the MetS.
The microcirculation dysfunction is a well-known effect of T2DM and the high prevalence of the subclinical neuropathy on diagnosis is a solid argument for the deleterious effects of the long-term preexisting insulin resistance.
Inside the endothelial cells, the insulin resistance generates an imbalance between the phosphatidylinositol-3-kinase (PI3-k) and the mitogen-activated protein kinase (MAPK) pathways which results in impaired vasodilatation, a procoagulant status, the NADPH-oxidase activation, and ROS production, smooth muscle cells proliferation, augmented response to catecholamines and endothelial dysfunction [41][42][43]. In CTS, the arteriolosclerosis of the small arteries of flexor tenosynovium was observed. The narrowing of the arteriolar lumen was due to the intimal hyperplasia related to the higher expression of matrix-metalloproteinases 2 (MMP-2) [44]. A similar vascular remodeling can be induced by insulin resistance. It has been demonstrated that diet-induced insulin resistance in rats increases MMP-2 arteriolar activity, a process which was reversed by doxycycline, a blocker of the activity site of MMP-2 [45].
In a hyperglycemic status, the advanced glycation end products (AGE) are formed in high amounts, the specific but not the only ligand of the advanced glycation end products receptor (RAGE). Experimental data showed that “advanced oxidation protein products”, food-derived advanced glycated end products, calgranulin, amphoterin, and amyloid-b-peptide released during metabolic or oxidative cellular stress, are also able to activate RAGE. The RAGE-induced signal activates nuclear factor-kB (Nf-KB), the early growth peptide 1, and the NADPH-oxidase and yields oxidative stress, inflammatory and prothrombotic species in atherosclerosis-prone vessels [41].
Hyperglycemia also influences lipid metabolism. The irreversible glycation of the LDL-C enhances its oxidative and inflammatory potential on the vascular cells [46]. Dyslipidemia generated by the enhanced lipolysis in adipocytes and de novo lipid synthesis in the hepatocytes increases the formation of the asymmetrical dimethylarginine (ADMA). ADMA inhibits the NO-synthetase, reducing the formation of nitric oxide (NO) and the vascular homeostasis [47]. Indeed, the ADMA was elevated in plasma from patients with MetS compared to controls [48].
MetS and hypothyroidism also share some common pathological mechanisms referring to vascular impairments such as the abnormal NO and vascular endothelial growth factor (VEGF) production [49]. A meta-analysis showed that VEGF-A is associated with diabetes, while VEGF-B and C are associated with the MetS and its components [50]. In vitro, the VEGF transcription is stimulated by TSH [51]. The members of the VEGF family have pleiotropic effects in vessels; they increase the permeability, which leads to edema and further narrowing of the CT space, stimulate the multiplication of the endothelial cells with intimal thickening, the development of collateral branches and the neovascularization [52] [53]. The neovascularization in sub-synovial connective tissue supports the high proliferation and thickening of the tendon sheet, further contributing to the narrowing of the tunnel which is more pronounced in diabetes CTS than in patients without T2DM [54]. The process seems to be influenced by the polymorphisms of the VEGF gene, as reflected by the more frequent neuropathy in patients with T2DM and D allele of the VEGF gene [55]. Leptin, an adipokine secreted in high levels by the visceral fat, has synergic effects with VEGF, on the capillary fenestration and permeability [56] [57].
The higher incidence of CTS in smokers and the relation with the MetS calls for some specific comments. The odds ratio of CTS in smokers is 4.862 (95% CI, 3.991-5.925) and many studies revealed an association between smoking and central obesity, even if there is no unanimous agreement [58][59][60][61][62]. Both body weight variation and nicotine addiction have genetic components [63][64]. In a large sample of current, former, and never smokers, investigators using the Genome-wide Complex Trait Analysis found a positive relationship between genetic factors influencing the smoking habit and BMI [65]. In this study, common genetic variants predisposing to the intensity of smoking behavior and an increased BMI were identified. The influence of gender on this association is also inconsistent, with studies in which the influence of smoking is predominated in women, and others in which it was restricted only to men [61][66]. Although both smoking and the female sex are well-known risk factors for CTS the interaction of these two is neglected, as it is for other pathologies [67][68]. At least from the biological perspective, prospective studies to capture the potential synergic effect of these risk factors are still needed.
In what concerns the pathological mechanism, smoking has multiple negative effects on the vascular system. Active smokers have thicker arterial walls, lower flow-mediated dilatation, and response to nitroglycerine [69]. Nicotine activates the sympathetic nervous system and the production of ROS and nitrogen species increases lipolysis in white adipose tissue and contributes to insulin resistance in muscle cells [70]. Nicotine also reduces the antioxidant enzymes (superoxide dismutase) and the activity of the endothelial nitric oxide synthase, altering the endothelial function [71]. Experimental data show a slow recovery after an ischemia-reperfusion injury related to smoking. [72]. In healthy non-smokers, a 30 minutes passive exposure to smoking decreases the rate of oxygen consumption and the reperfusion after vascular occlusion and the capillary blood flow is significantly decreased[73][74]. In experiments conducted on the extensor digitorum longus, the chronic adaptation to hypoxia related to the blockade of hemoglobin with carbon monoxide and vasoconstriction diminished the volume of the type II muscle fibers and increased the fiber oxidative enzyme activity [75].
The relation between MetS, cardiovascular diseases, and CTS is also endorsed by epidemiological data. Hypertensive patients aged 30-44 from a Finish population-based survey, had an OR of 3.4, (95% CI 1.6-7.4) for CTS; the association with cardiac arrhythmia was even stronger (OR 10.2, 95% CI 2.7-38.4) [76]. The relation between high blood pressure and CTS was maintained after adjustment to gender, BMI, and occupational risk factors, expressed in the strain index [77]. It is also of interest that CTS might predict future risk of coronary heart disease cardiac failure, atrial fibrillation, atrioventricular heart block, and pacemaker implantation [78][79]. Most probably due to the shared risk factors (dyslipidemia, impaired metabolism and inflammation) and the effects of the MetS on the microvascular structure which lead to a reduction in the endoneurial blood flow, oxygenation, and hypoxia of the median nerve.
This entry is adapted from the peer-reviewed paper 10.3390/cimb44060181
References
- Rens Hanewinckel; Judith Drenthen; Symen Ligthart; Abbas Dehghan; Oscar Franco; Albert Hofman; M Arfan Ikram; Pieter A Van Doorn; Metabolic syndrome is related to polyneuropathy and impaired peripheral nerve function: a prospective population-based cohort study. Journal of Neurology, Neurosurgery & Psychiatry 2016, 87, 1336-1342, 10.1136/jnnp-2016-314171.
- A. Gordon Smith; J. Robinson Singleton; Idiopathic neuropathy, prediabetes and the metabolic syndrome. Journal of the Neurological Sciences 2006, 242, 9-14, 10.1016/j.jns.2005.11.020.
- Ling Han; Lijin Ji; Jing Chang; Jian Wen; Wenting Zhao; Hongli Shi; Linuo Zhou; Yiming Li; Renming Hu; Ji Hu; et al. Peripheral neuropathy is associated with insulin resistance independent of metabolic syndrome. Diabetology & Metabolic Syndrome 2015, 7, 14-14, 10.1186/s13098-015-0010-y.
- Eugenia Rota; Roberto Quadri; Edoardo Fanti; Gianluca Isoardo; Fabio Poglio; Alessia Tavella; Ilaria Paolasso; Palma Ciaramitaro; Bruno Bergamasco; Dario Cocito; et al. Electrophysiological findings of peripheral neuropathy in newly diagnosed type II diabetes mellitus. Journal of the Peripheral Nervous System 2005, 10, 348-353, 10.1111/j.1085-9489.2005.00046.x.
- Sonoko Misawa; Satoshi Kuwabara; Kazue Ogawara; Yukiko Kitano; Kazuo Yagui; Takamichi Hattori; Hyperglycemia alters refractory periods in human diabetic neuropathy. Clinical Neurophysiology 2004, 115, 2525-2529, 10.1016/j.clinph.2004.06.008.
- Nádia P. Gonçalves; Christian B. Vægter; Henning Andersen; Leif Østergaard; Nigel A. Calcutt; Troels S. Jensen; Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nature Reviews Neurology 2017, 13, 135-147, 10.1038/nrneurol.2016.201.
- Srikanth, K.K.; Orrick, J.A. Polyol Or Sorbitol Pathways; StatPearls : [Internet], 2022; pp. www.ncbi.nlm.nih.gov/books/NBK576381.
- Claudia Figueroa-Romero; Mahdieh Sadidi; Eva L. Feldman; Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Reviews in Endocrine and Metabolic Disorders 2008, 9, 301-314, 10.1007/s11154-008-9104-2.
- Wu Hao; Syoichi Tashiro; Tomoka Hasegawa; Yuiko Sato; Tami Kobayashi; Toshimi Tando; Eri Katsuyama; Atsuhiro Fujie; Ryuichi Watanabe; Mayu Morita; et al. Hyperglycemia Promotes Schwann Cell De-differentiation and De-myelination via Sorbitol Accumulation and Igf1 Protein Down-regulation. Journal of Biological Chemistry 2015, 290, 17106-17115, 10.1074/jbc.m114.631291.
- Hania S Kassem; Sami T Azar; Mira S Zantout; Raja A Sawaya; Hypertriglyceridemia and peripheral neuropathy in neurologically asymptomatic patients.. Neuro endocrinology letters 2005, 26, 775-9, .
- Timothy D. Wiggin; Kelli A. Sullivan; Rodica Pop-Busui; Antonino Amato; Anders A.F. Sima; Eva L. Feldman; Elevated Triglycerides Correlate With Progression of Diabetic Neuropathy. Diabetes 2009, 58, 1634-1640, 10.2337/db08-1771.
- H.H. Beyca; B. Mesci; O. Telci Caklili; H.H. Mutlu; A. Oguz; Neuropathy Associated with Hypertriglyceridemia in Patients with Metabolic Syndrome. Acta Endocrinologica (Bucharest) 2016, 12, 26-29, 10.4183/aeb.2016.26.
- Natalie C. G. Kwai; William Nigole; Ann M. Poynten; Chris Brown; Arun V. Krishnan; The Relationship between Dyslipidemia and Acute Axonal Function in Type 2 Diabetes Mellitus In Vivo. PLOS ONE 2016, 11, e0153389, 10.1371/journal.pone.0153389.
- Zixin Cai; Yan Yang; Jingjing Zhang; A systematic review and meta-analysis of the serum lipid profile in prediction of diabetic neuropathy. Scientific Reports 2021, 11, 1-20, 10.1038/s41598-020-79276-0.
- Edward K. Duran; Aaron Aday; Nancy R. Cook; Julie E. Buring; Paul M Ridker; Aruna D. Pradhan; Triglyceride-Rich Lipoprotein Cholesterol, Small Dense LDL Cholesterol, and Incident Cardiovascular Disease. Journal of the American College of Cardiology 2020, 75, 2122-2135, 10.1016/j.jacc.2020.02.059.
- Sergey Lupachyk; Pierre Watcho; Nailia Hasanova; Ulrich Julius; Irina G. Obrosova; Triglyceride, nonesterified fatty acids, and prediabetic neuropathy: role for oxidative–nitrosative stress. Free Radical Biology and Medicine 2012, 52, 1255-1263, 10.1016/j.freeradbiomed.2012.01.029.
- Sergey Lupachyk; Pierre Watcho; Nailia Hasanova; Ulrich Julius; Irina G. Obrosova; Triglyceride, nonesterified fatty acids, and prediabetic neuropathy: role for oxidative–nitrosative stress. Free Radical Biology and Medicine 2012, 52, 1255-1263, 10.1016/j.freeradbiomed.2012.01.029.
- J F Poduslo; G L Curran; Increased permeability across the blood-nerve barrier of albumin glycated in vitro and in vivo from patients with diabetic polyneuropathy.. Proceedings of the National Academy of Sciences 1992, 89, 2218-2222, 10.1073/pnas.89.6.2218.
- Philip J. Barter; Stephen Nicholls; Kerry-Anne Rye; G. M. Anantharamaiah; Mohamad Navab; Alan M. Fogelman; Antiinflammatory Properties of HDL. Circulation Research 2004, 95, 764-772, 10.1161/01.res.0000146094.59640.13.
- Paul Holvoet; Association Between Circulating Oxidized Low-Density Lipoprotein and Incidence of the Metabolic Syndrome. JAMA 2008, 299, 2287-2293, 10.1001/jama.299.19.2287.
- Andrea M. Vincent; John M. Hayes; Lisa L. McLean; Anuradha Vivekanandan-Giri; Subramaniam Pennathur; Eva L. Feldman; Dyslipidemia-Induced Neuropathy in Mice. Diabetes 2009, 58, 2376-2385, 10.2337/db09-0047.
- Eun-Ra Jang; Chung Soo Lee; 7-Ketocholesterol induces apoptosis in differentiated PC12 cells via reactive oxygen species-dependent activation of NF-κB and Akt pathways. Neurochemistry International 2011, 58, 52-59, 10.1016/j.neuint.2010.10.012.
- Bo Wang; Peter Tontonoz; Liver X receptors in lipid signalling and membrane homeostasis. Nature Reviews Endocrinology 2018, 14, 452-463, 10.1038/s41574-018-0037-x.
- Chaitanya K. Gavini; RaizA Bonomo; Virginie Mansuy-Aubert; Neuronal LXR Regulates Neuregulin 1 Expression and Sciatic Nerve-Associated Cell Signaling in Western Diet-fed Rodents. Scientific Reports 2020, 10, 1-12, 10.1038/s41598-020-63357-1.
- Chaitanya K. Gavini; Nadia Elshareif; Gregory Aubert; Anand V. Germanwala; Nigel A. Calcutt; Virginie Mansuy-Aubert; LXR agonist improves peripheral neuropathy and modifies PNS immune cells in aged mice. Journal of Neuroinflammation 2022, 19, 1-14, 10.1186/s12974-022-02423-z.
- Rahman Shiri; Hypothyroidism and carpal tunnel syndrome: A meta-analysis. Muscle & Nerve 2014, 50, 879-883, 10.1002/mus.24453.
- Faris Aldaghri; Mohammed S. Algahtani; Talal A. Almutairi; Moath Albusair; Khalid Bin Ghali; Fahad S. Al Asim; Prevalence of Hypothyroidism Among Carpal Tunnel Syndrome Patients at a Hospital in Saudi Arabia. Cureus 2020, 12, e12264, 10.7759/cureus.12264.
- Sampada Swapneel Karne; Carpal Tunnel Syndrome in Hypothyroidism. JOURNAL OF CLINICAL AND DIAGNOSTIC RESEARCH 2016, 10, OC36-8, 10.7860/jcdr/2016/16464.7316.
- Stephan Ruhla; Martin O. Weickert; Ayman Arafat; Martin Osterhoff; Frank Isken; Joachim Spranger; Christof Schöfl; Andreas F. H. Pfeiffer; Matthias Möhlig; A high normal TSH is associated with the metabolic syndrome. Clinical Endocrinology 2010, 72, 696-701, 10.1111/j.1365-2265.2009.03698.x.
- Leonidas Duntas; Gabriela Brenta; A Renewed Focus on the Association Between Thyroid Hormones and Lipid Metabolism. Frontiers in Endocrinology 2018, 9, 511, 10.3389/fendo.2018.00511.
- Aysin Oge; E. Sozmen; A. O. Karaoglu; Effect of Thyroid Function on LDL Oxidation in Hypothyroidism and Hyperthyroidism. Endocrine Research 2004, 30, 481-489, 10.1081/erc-200036185.
- Xavier Prieur; Thierry Huby; Hervé Coste; Frank Schaap; M. John Chapman; Joan C. Rodríguez; Thyroid Hormone Regulates the Hypotriglyceridemic Gene APOA5. Journal of Biological Chemistry 2005, 280, 27533-27543, 10.1074/jbc.m503139200.
- Fabrizio Damiano; Alessio Rochira; Antonio Gnoni; Luisa Siculella; Action of Thyroid Hormones, T3 and T2, on Hepatic Fatty Acids: Differences in Metabolic Effects and Molecular Mechanisms. International Journal of Molecular Sciences 2017, 18, 744, 10.3390/ijms18040744.
- Annemarie Gagnon; Tayze T. Antunes; Tapraya Ly; Patama Pongsuwan; Claire Gavin; Heather A. Lochnan; Alexander Sorisky; Thyroid-stimulating hormone stimulates lipolysis in adipocytes in culture and raises serum free fatty acid levels in vivo. Metabolism 2010, 59, 547-553, 10.1016/j.metabol.2009.08.018.
- Lindsey R. Boone; William Lagor; Margarita De La Llera Moya; Melissa I. Niesen; George H. Rothblat; Gene C. Ness; Thyroid hormone enhances the ability of serum to accept cellular cholesterol via the ABCA1 transporter. Atherosclerosis 2011, 218, 77-82, 10.1016/j.atherosclerosis.2011.04.028.
- Huixing Liu; Daoquan Peng; Update on dyslipidemia in hypothyroidism: the mechanism of dyslipidemia in hypothyroidism. Endocrine Connections 2022, 11, e210002, 10.1530/ec-21-0002.
- Dana Holovacova; Martin Kužma; Zdenko Killinger; Juraj Payer; Cross-sectional area of the median nerve is increased in primary autoimmune hypothyroidism and decreases upon treatment with thyroxine. European Journal of Endocrinology 2016, 175, 265-271, 10.1530/eje-16-0397.
- L Perbellini; P Mozzo; F Brugnone; A Zedde; Physiologicomathematical model for studying human exposure to organic solvents: kinetics of blood/tissue n-hexane concentrations and of 2,5-hexanedione in urine.. Occupational and Environmental Medicine 1986, 43, 760-768, 10.1136/oem.43.11.760.
- Richard M. LoPachin; Terrence Gavin; Toxic neuropathies: Mechanistic insights based on a chemical perspective. Neuroscience Letters 2014, 596, 78-83, 10.1016/j.neulet.2014.08.054.
- M. Otelea; C. Handra; A. Rascu; Registered cases of occupational n-hexane intoxication in Bucharest. Romanian Journal of Legal Medicine 2015, 23, 279-284, 10.4323/rjlm.2015.279.
- Brian H. Cohen; Michael Gaspar; Alan H. Daniels; Edward Akelman; Patrick M. Kane; Multifocal Neuropathy: Expanding the Scope of Double Crush Syndrome. The Journal of Hand Surgery 2016, 41, 1171-1175, 10.1016/j.jhsa.2016.09.009.
- Joseph Loscalzo; Richard C Jin; Vascular nitric oxide: formation and function. Journal of Blood Medicine 2010, 1, 147-162, 10.2147/jbm.s7000.
- Gerhard H. Scholz; Markolf Hanefeld; Metabolic Vascular Syndrome: New Insights into a Multidimensional Network of Risk Factors and Diseases. Visceral Medicine 2016, 32, 319-326, 10.1159/000450866.
- Alejandro Gutiérrez; Cristina Contreras; Ana Sánchez; Dolores Prieto; Role of Phosphatidylinositol 3-Kinase (PI3K), Mitogen-Activated Protein Kinase (MAPK), and Protein Kinase C (PKC) in Calcium Signaling Pathways Linked to the α1-Adrenoceptor in Resistance Arteries. Frontiers in Physiology 2019, 10, 55, 10.3389/fphys.2019.00055.
- Hitoshi Hirata; Masaya Tsujii; Toshimichi Yoshida; Kyoko Imanaka Yoshida; Akimasa Morita; Noritaka Okuyama; Takeshi Nagakura; Toshiko Sugimoto; Kohzo Fujisawa; Atsumasa Uchida; et al. MMP-2 expression is associated with rapidly proliferative arteriosclerosis in the flexor tenosynovium and pain severity in carpal tunnel syndrome. The Journal of Pathology 2005, 205, 443-450, 10.1002/path.1709.
- Pr Nagareddy; Ps Rajput; H Vasudevan; B McClure; U Kumar; Km MacLeod; Jh McNeill; Inhibition of matrix metalloproteinase-2 improves endothelial function and prevents hypertension in insulin-resistant rats. British Journal of Pharmacology 2011, 165, 705-715, 10.1111/j.1476-5381.2011.01583.x.
- Laura Toma; Camelia Sorina Stancu; Gabriela M. Botez; Anca V. Sima; Maya Simionescu; Irreversibly glycated LDL induce oxidative and inflammatory state in human endothelial cells; added effect of high glucose. Biochemical and Biophysical Research Communications 2009, 390, 877-882, 10.1016/j.bbrc.2009.10.066.
- Lorna Fiedler; The DDAH/ADMA pathway is a critical regulator of NO signalling in vascular homeostasis. Cell Adhesion & Migration 2008, 2, 149-150, 10.4161/cam.2.3.6819.
- Iván Palomo; Alejandra Contreras; Marcelo Alarcon; Elba Leiva; Luis Guzman; Veronica Mujica; Gloria Icaza; Nora Díaz; Daniel Gonzalez; Rodrigo Moore-Carrasco; et al. Elevated concentration of asymmetric dimethylarginine (ADMA) in individuals with metabolic syndrome. Nitric Oxide 2011, 24, 224-228, 10.1016/j.niox.2011.03.002.
- Zoran Gluvic; Milan M. Obradovic; Emina M. Sudar-Milovanovic; Sonja S. Zafirovic; Djordje J. Radak; Magbubah M. Essack; Vladimir B. Bajic; Gojobori Takashi; Esma R. Isenovic; Regulation of nitric oxide production in hypothyroidism. Biomedicine & Pharmacotherapy 2020, 124, 109881, 10.1016/j.biopha.2020.109881.
- Mohammad Ishraq Zafar; Kerry Mills; Xiaofeng Ye; Brette Blakely; Jie Min; Wen Kong; Nan Zhang; Luoning Gou; Anita Regmi; Sheng Qing Hu; et al. Association between the expression of vascular endothelial growth factors and metabolic syndrome or its components: a systematic review and meta-analysis.. Diabetology & Metabolic Syndrome 2018, 10, 62, 10.1186/s13098-018-0363-0.
- Alejandro Gutiérrez; Cristina Contreras; Ana Sánchez; Dolores Prieto; Role of Phosphatidylinositol 3-Kinase (PI3K), Mitogen-Activated Protein Kinase (MAPK), and Protein Kinase C (PKC) in Calcium Signaling Pathways Linked to the α1-Adrenoceptor in Resistance Arteries. Frontiers in Physiology 2019, 10, 55, 10.3389/fphys.2019.00055.
- Sebastian Hoffmann; Lorenz C. Hofbauer; Vera Scharrenbach; Anette Wunderlich; Iyad Hassan; Susanne Lingelbach; Andreas Zielke; Thyrotropin (TSH)-Induced Production of Vascular Endothelial Growth Factor in Thyroid Cancer Cellsin Vitro: Evaluation of TSH Signal Transduction and of Angiogenesis-Stimulating Growth Factors. The Journal of Clinical Endocrinology & Metabolism 2004, 89, 6139-6145, 10.1210/jc.2004-1260.
- A N Deger; H Deger; F Taser; The role of neoangiogenesis and vascular endothelial growth factor in the development of carpal tunnel syndrome in patients with diabetes. Nigerian Journal of Clinical Practice 2016, 19, 189-95, 10.4103/1119-3077.175971.
- G. Donato; Olimpio Galasso; P. Valentino; F. Conforti; V. Zuccalà; Emilio Russo; L. Maltese; Ida Perrotta; Sandro Tripepi; A. Amorosi; et al. Pathological findings in subsynovial connective tissue in idiopathic carpal tunnel syndrome. Clinical Neuropathology 2009, 28, 129-135, 10.5414/npp28129.
- Adina Stoian; Anca Bacârea; Anca Motataianu; Mircea Stoian; Florina Gliga; Vladimir Bacârea; Carmen Duicu; Claudia Bănescu; Vascular Endothelial Growth Factor Insertion/Deletion gene polymorphism in patients with type 2 diabetes and diabetic peripheral polyneuropathy. Revista Romana de Medicina de Laborator 2014, 22, 165-172, 10.2478/rrlm-2014-0023.
- V Van Harmelen; S Reynisdottir; P Eriksson; A Thörne; J Hoffstedt; F Lönnqvist; P Arner; Leptin secretion from subcutaneous and visceral adipose tissue in women.. Diabetes 1998, 47, 913-917, 10.2337/diabetes.47.6.913.
- Renhai Cao; Ebba Brakenhielm; Claes Wahlestedt; Johan Thyberg; Yihai Cao; Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proceedings of the National Academy of Sciences 2001, 98, 6390-6395, 10.1073/pnas.101564798.
- Wenjie Guan; Jie Lao; Yudong Gu; Xin Zhao; Jing Rui; Kaiming Gao; Case-control study on individual risk factors of carpal tunnel syndrome. Experimental and Therapeutic Medicine 2018, 15, 2761-2766, 10.3892/etm.2018.5817.
- Yeonjung Kim; Seong Min Jeong; Bora Yoo; Bitna Oh; Hee-Cheol Kang; Associations of smoking with overall obesity, and central obesity: a cross-sectional study from the Korea National Health and Nutrition Examination Survey (2010-2013). Epidemiology and Health 2016, 38, e2016020, 10.4178/epih.e2016020.
- Sidsel Graff-Iversen; Stephen Hewitt; Lisa Forsén; Liv Grøtvedt; Inger Ariansen; Associations of tobacco smoking with body mass distribution; a population-based study of 65,875 men and women in midlife.. BMC Public Health 2019, 19, 1439-10, 10.1186/s12889-019-7807-9.
- Jun Lv; Wei Chen; Dianjianyi Sun; Shengxu Li; Iona Y. Millwood; Margaret Smith; Yu Guo; Zheng Bian; Canqing Yu; Huiyan Zhou; et al. Gender-Specific Association between Tobacco Smoking and Central Obesity among 0.5 Million Chinese People: The China Kadoorie Biobank Study. PLOS ONE 2015, 10, e0124586, 10.1371/journal.pone.0124586.
- Fernando Lanas; Lydia Bazzano; Adolfo Rubinstein; Matias Calandrelli; Chung-Shiuan Chen; Natalia Elorriaga; Laura Gutierrez; Jose A. Manfredi; Pamela Seron; Nora Mores; et al. Prevalence, Distributions and Determinants of Obesity and Central Obesity in the Southern Cone of America. PLOS ONE 2016, 11, e0163727, 10.1371/journal.pone.0163727.
- Ruth J. F. Loos; Giles S. H. Yeo; The genetics of obesity: from discovery to biology. Nature Reviews Genetics 2021, 23, 120-133, 10.1038/s41576-021-00414-z.
- Chirila, M.; Ghita, I.; Handra C.; Fulga, I; Genetic variants influencing smoking behavior and efficacy of smoking cessation therapies. Rom Biotechnol Lett 2014, 19 (5), 9727-9734, .
- Amanda G. Wills; Christian Hopfer; Phenotypic and genetic relationship between BMI and cigarette smoking in a sample of UK adults. Addictive Behaviors 2018, 89, 98-103, 10.1016/j.addbeh.2018.09.025.
- Eeva-Liisa Tuovinen; Suoma E. Saarni; Satu Männistö; Katja Borodulin; Kristiina Patja; Taru H. Kinnunen; Jaakko Kaprio; Tellervo Korhonen; Smoking status and abdominal obesity among normal- and overweight/obese adults: Population-based FINRISK study. Preventive Medicine Reports 2016, 4, 324-330, 10.1016/j.pmedr.2016.07.003.
- Sina Hulkkonen; Rahman Shiri; Juha Auvinen; Jouko Miettunen; Jaro Karppinen; Jorma Ryhänen; Risk factors of hospitalization for carpal tunnel syndrome among the general working population. Scandinavian Journal of Work, Environment & Health 2019, 46, 43-49, 10.5271/sjweh.3835.
- Florin Dumitru Mihaltan; Armand-Gabriel Rajnoveanu; Ruxandra-Mioara Rajnoveanu; Impact of Smoking on Women during the Covid-19 Pandemic. null 2021, 8, 584061, 10.37247/pamed2ed.3.2021.17.
- Ali Metin Esen; Irfan Barutcu; Murat Acar; Bumin Degirmenci; Dayimi Kaya; Muhsin Turkmen; Mehmet Melek; Ersel Onrat; Ozlem Batukan Esen; Cevat Kirma; et al. Effect of Smoking on Endothelial Function and Wall Thickness of Brachial Artery. Circulation Journal 2004, 68, 1123-1126, 10.1253/circj.68.1123.
- Xi-Yong Yu; Ping Song; Ming-Hui Zou; Obesity Paradox and Smoking Gun. Circulation Research 2018, 122, 1642-1644, 10.1161/circresaha.118.312897.
- Hong-Li Luo; Wei-Jin Zang; Jun Lu; Xiao-Jiang Yu; Yuan-Xi Lin; Yong-Xiao Cao; The Protective Effect of Captopril on Nicotine-Induced Endothelial Dysfunction in Rat. Basic & Clinical Pharmacology & Toxicology 2006, 99, 237-245, 10.1111/j.1742-7843.2006.pto_494.x.
- Brian Rinker; Betsy F. Fink; Neil G. Barry; Joshua A. Fife; Maria E. Milan; Ashley R. Stoker; Peter T. Nelson; The effect of cigarette smoking on functional recovery following peripheral nerve ischemia/reperfusion injury. Microsurgery 2010, 31, 59-65, 10.1002/micr.20820.
- V. Linardatou; E. Karatzanos; N. Panagopoulou; D. Delis; C. Kourek; N. Rovina; S. Nanas; I. Vasileiadis; Passive smoking acutely affects the microcirculation in healthy non-smokers. Microvascular Research 2019, 128, 103932, 10.1016/j.mvr.2019.103932.
- Peter Henriksson; Qing Lu; Ulf Diczfalusy; Anna Freyschuss; Immediate Effect of Passive Smoking on Microcirculatory Flow. Microcirculation 2014, 21, 587-592, 10.1111/micc.12137.
- Toshiaki Nakatani; Toshikatsu Nakashima; Taizo Kita; Akihiko Ishihara; Effects of exposure to cigarette smoke at different dose levels on extensor digitorum longus muscle fibres in Wistar-Kyoto and spontaneously hypertensive rats. Clinical and Experimental Pharmacology and Physiology 2003, 30, 671-677, 10.1046/j.1440-1681.2003.03898.x.
- Rahman Shiri; Markku Heliövaara; Leena Moilanen; Jorma Viikari; Helena Liira; Eira Viikari-Juntura; Associations of cardiovascular risk factors, carotid intima-media thickness and manifest atherosclerotic vascular disease with carpal tunnel syndrome. BMC Musculoskeletal Disorders 2011, 12, 80-80, 10.1186/1471-2474-12-80.
- Kurt T. Hegmann; Matthew Steven Thiese; Jay Kapellusch; Andrew S. Merryweather; Stephen Bao; Barbara Silverstein; Eric M. Wood; Richard Kendall; Jacqueline Wertsch; James Foster; et al. Association Between Cardiovascular Risk Factors and Carpal Tunnel Syndrome in Pooled Occupational Cohorts. Journal of Occupational & Environmental Medicine 2016, 58, 87-93, 10.1097/jom.0000000000000573.
- Yi-Chuan Chang; Jen-Huai Chiang; Ing-Shiow Lay; Yu-Chen Lee; Increased Risk of Coronary Artery Disease in People with a Previous Diagnosis of Carpal Tunnel Syndrome: A Nationwide Retrospective Population-Based Case-Control Study. BioMed Research International 2019, 2019, 1-8, 10.1155/2019/3171925.