Mycotoxins are a group of natural compounds produced under a wide range of climatic conditions by filamentous fungi mainly belonging to Aspergillus, Penicillium, Fusarium, Claviceps and Alternaria species. These toxins can contaminate various agricultural commodities (cereals, dried fruits, nuts, spices and coffee being the most frequently contaminated ones) either before harvest or under postharvest conditions, thus posing a risk to human and animal health due to their toxic effects.
- mycotoxins
- food
- LC-MS methods
1. Introduction
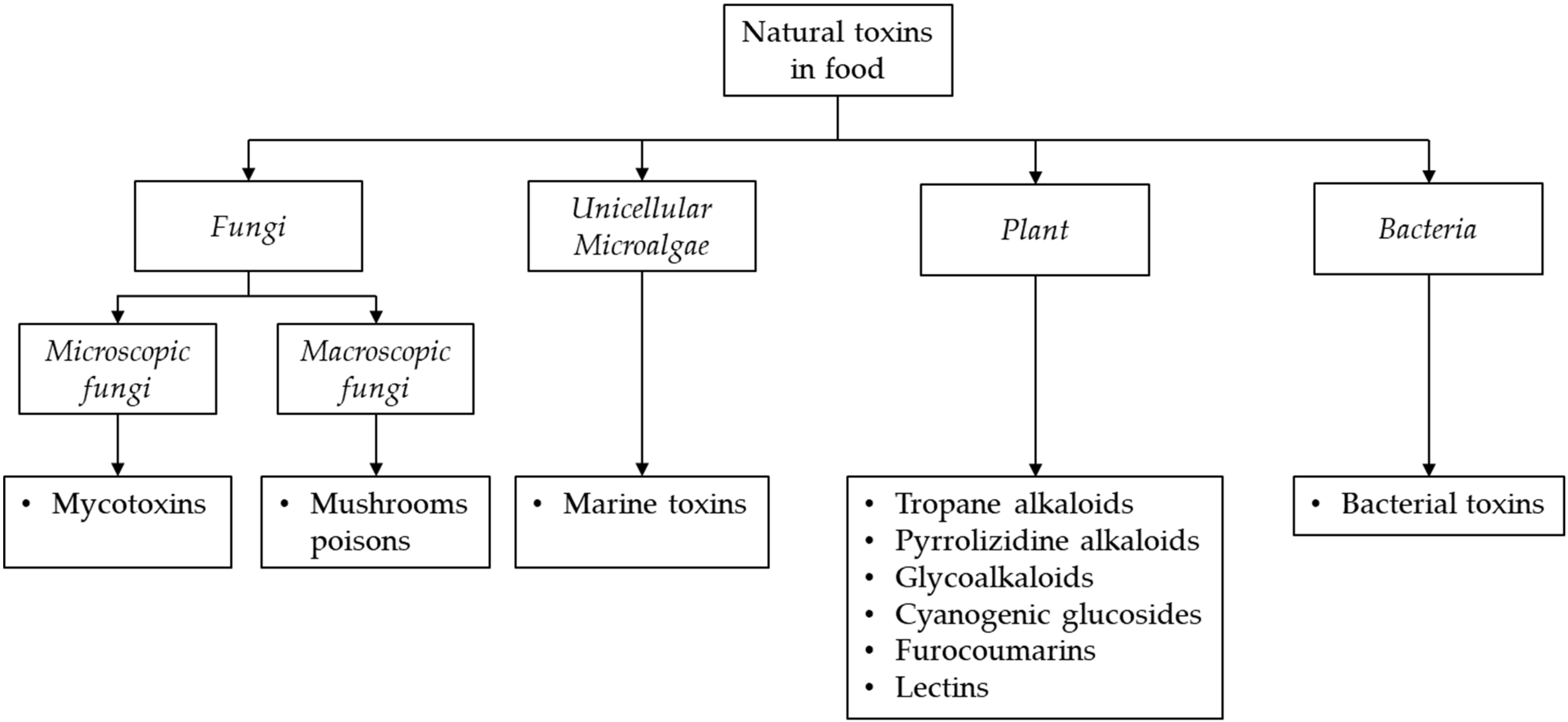
The reduction of risks related to the presence of natural toxins in food plays an essential role in protecting consumers. Indeed, WHO, together with the European Food Safety Authority (EFSA), FAO and Codex Alimentarius Commission, have established maximum residue limits (MRLs) or recommendations for many of these natural toxins to control their occurrence in food [7,8,9,10,11,12,13,14]. Mycotoxins are one of the most relevant threats to human health. They represent the most important class of chemical hazards among natural toxins recorded in the European Rapid Alert System on Feed and Food (RASFF). To ensure a correct risk assessment evaluation and compliance with the current legislation, it is important to develop sensitive, selective and robust analytical methods to determine the occurrence of these class of natural toxins in foodstuffs. However, the main challenge for the analysis of these compounds in food samples is related to their different physico-chemical properties, the inherent complexity of food matrices as well as the purpose of the analysis. These characteristics can affect the extraction efficiency of targeted toxins and then the accuracy and sensitivity of the method [4].
2. Mycotoxins
The scientific progress in the application of LC-MS-based methods to the analysis of mycotoxins in food, considering the literature published from 2016 to 2021 is reported below. The literature search employed the Scopus online database (www.scopus.com, accessed on 31 January 2022) and keywords used for the search were “mycotoxins”, “liquid chromatography mass spectrometry” and “food. Several papers were published in the selective period with a positive trend over the years. A further refinement of selected papers was applied in order to select only those describing the development and validation of an LC-MS method (together with performance characteristics values). The novelty of the used technology, the application to new food matrices and the possibility to simultaneously analyze multiple toxins also including emerging ones were taken into account for the selection.
3. LC-MS Methods for Mycotoxin Determination in Food
This entry is adapted from the peer-reviewed paper 10.3390/toxins14050328
References
- WHO Natural Toxins in food (Factsheet). Available online: www.who.int/news-room/fact-sheets/detail/natural-toxins-in-food (accessed on 5 April 2022).
- Dolan, L.C.; Matulka, R.A.; Burdock, G.A. Naturally Occurring Food Toxins. Toxins 2010, 2, 2289–2332.
- Wong, Y.; Lewis, R.J. Analysis of Food Toxins and Toxicants; Wong, Y.C., Lewis, R.J., Eds.; John Wiley & Sons: Chichester, UK, 2017; Volumes 1–2.
- Casado, N.; Gañán, J.; Morante-Zarcero, S.; Sierra, I. New Advanced Materials and Sorbent-Based Microextraction Techniques as Strategies in Sample Preparation to Improve the Determination of Natural Toxins in Food Samples. Molecules 2020, 25, 702.
- Hajslova, J.; Schulzova, V.; Botek, P.; Lojza, J. Natural toxins in food crops and their changes during processing. Czech J. Food Sci. 2004, 22, S29–S34.
- Rodríguez, I.; González, J.M.; Botana, A.M.; Sainz, M.J.; Vieytes, M.R.; Alfonso, A.; Botana, L.M. Analysis of natural toxins by liquid chromatography. In Liquid Chromatography, 2nd ed.; Fanali, S., Haddad, P.R., Poole, C.F., Riekkola, M.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 479–514.
- Creppy, E.E. Update of survey, regulation and toxic effects of mycotoxins in Europe. Toxicol. Lett. 2002, 127, 19–28.
- IARC International Agency for Research on Cancer. Some Industrial Chemical Intermediates; International Agency for Research on Cancer: Lyon, France, 2020; Volume 125, ISBN 9789283201649.
- European Commission. Commission Regulation (EC) No. 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, L364, 5–24.
- European Commission. Commission Regulation (EU) No. 2016/239 of 19 February 2016 amending Regulation (EC) No 1881/2006 as regards maximum levels of tropane alkaloids in certain cereal-based foods for infants and young children. Off. J. Eur. Union 2016, L45, 3–5.
- European Commission. Commission Regulation (EC) No. 1126/2007 of 28 September 2007 amending Regulation (EC) No. 1881/2006 setting maximum levels for certain contaminants in foodstuffs as regards Fusarium toxins in maize and maize products. Off. J. Eur. Union 2007, L255, 14–17.
- European Commission. Commission Recommendation of 27 March 2013 on the presence of T-2 and HT-2 toxin in cereals and cereal products (2013/165/EU). Off. J. Eur. Union 2013, L91, 12–15.
- European Commission. Commission Regulation (EU) No 212/2014 of 6 March 2014 amending Regulation (EC) No. 1881/2006 as regards maximum levels of the contaminant citrinin in food supplements based on rice fermented with red yeast Monascus purpureus. Off. J. Eur. Union 2014, L67, 3–4.
- EFSA (European Food Safety Authority). Evaluation of the health risks related to the presence of cyanogenic glycosides in foods other than raw apricot kernels. EFSA J. 2019, 17, e05662.
- EFSA (European Food Safety Authority). Human and animal dietary exposure to ergot alkaloids European. EFSA J. 2017, 15, e04902.
- EFSA (European Food Safety Authority). Dietary exposure assessment to Alternaria toxins in the European population. EFSA J. 2016, 14, e04654.
- Eskola, M.; Elliott, C.T.; Hajslova, J.; Steiner, D.; Krska, R. Towards a dietary-exposome assessment of chemicals in food: An update on the chronic health risks for the European consumer. Crit. Rev. Food Sci. Nutr. 2019, 60, 1890–1911.
- Kovalsky, P.; Kos, G.; Nährer, K.; Schwab, C.; Jenkins, T.; Schatzmayr, G.; Sulyok, M.; Krska, R. Co-Occurrence of Regulated, Masked and Emerging Mycotoxins and Secondary Metabolites in Finished Feed and Maize—An Extensive Survey. Toxins 2016, 8, 363.
- EFSA (European Food Safety Authority). Scientific Opinion on the risks for animal and public health related to the presence of Alternariatoxins in feed and food. EFSA J. 2011, 9, 2407.
- EFSA (European Food Safety Authority). Scientific Opinion on the risk for public and animal health related to the presence of sterigmatocystin in food and feed. EFSA J. 2013, 11, 3254.
- EFSA (European Food Safety Authority). Scientific Opinion on risks for animal and public health related to the presence of nivalenol in food and feed. EFSA J. 2013, 11, 3262.
- EFSA (European Food Safety Authority). Scientific Opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 12, 3802.
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging mycotoxins: Beyond traditionally determined food contaminants. J. Agric. Food Chem. 2017, 65, 7052–7070.
- Pascale, M.; De Girolamo, A.; Lippolis, V.; Stroka, J.; Mol, H.; Lattanzio, V.M. Performance Evaluation of LC-MS Methods for Multimycotoxin Determination. J. AOAC Int. 2019, 102, 1708–1720.
- Leite, M.; Freitas, A.; Silva, A.S.; Barbosa, J.; Ramos, F. Maize (Zea mays L.) and mycotoxins: A review on optimization and validation of analytical methods by liquid chromatography coupled to mass spectrometry. Trends Food Sci. Technol. 2020, 99, 542–565.
- Medina, D.A.V.; Borsatto, J.V.B.; Maciel, E.V.S.; Lanças, F.M. Current role of modern chromatography and mass spectrometry in the analysis of mycotoxins in food. TrAC Trends Anal. Chem. 2021, 135, 116156.
- Malachová, A.; Stránská, M.; Václavíková, M.; Elliott, C.T.; Black, C.; Meneely, J.; Hajšlová, J.; Ezekiel, C.N.; Schuhmacher, R.; Krska, R. Advanced LC–MS-based methods to study the co-occurrence and metabolization of multiple mycotoxins in cereals and cereal-based food. Anal. Bioanal. Chem. 2017, 410, 801–825.
- Steiner, D.; Malachová, A.; Sulyok, M.; Krska, R. Challenges and future directions in LC-MS-based multiclass method development for the quantification of food contaminants. Anal. Bioanal. Chem. 2020, 413, 25–34.
- Righetti, L.; Paglia, G.; Galaverna, G.; Dall’Asta, C. Recent Advances and Future Challenges in Modified Mycotoxin Analysis: Why HRMS Has Become a Key Instrument in Food Contaminant Research. Toxins 2016, 8, 361.
- European Commission. Mandate for Standardisation Addressed to CEN for Methods of Analysis for Mycotoxins in Food 2013. Available online: https://law.resource.org/pub/eu/mandates/m520.pdf (accessed on 5 April 2022).
- Juan, C.; Covarelli, L.; Beccari, G.; Colasante, V.; Mañes, J. Simultaneous analysis of twenty-six mycotoxins in durum wheat grain from Italy. Food Control 2016, 62, 322–329.
- European Commission. Commission Regulation (EU) No 519/2014 of 16 May 2014 amending Regulation (EC) No. 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis. Off. J. Eur. Union 2014, L147, 29–43.
- Kai, S.; Kosuge, N.; Waki, M.; Miyazawa, M.; Kanazawa, H. Analysis of Fusarium Toxins in Processed Grain Products Using High-Performance Liquid Chromatography/Tandem Mass Spectrometry. Chromatography 2016, 37, 79–85.
- Sharmili, K.; Jinap, S.; Sukor, R. Development, optimization and validation of QuEChERS based liquid chromatography tandem mass spectrometry method for determination of multimycotoxin in vegetable oil. Food Control 2016, 70, 152–160.
- Sun, J.; Li, W.; Zhang, Y.; Hu, X.; Wu, L.; Wang, B. QuEChERS Purification Combined with Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry for Simultaneous Quantification of 25 Mycotoxins in Cereals. Toxins 2016, 8, 375.
- Wang, Y.; Dong, Y.-J.; Li, Z.-M.; Deng, L.-G.; Guo, C.-Y.; Zhang, S.-Q.; Li, D.-P.; Zhao, S.-C. Fast determination of multi-mycotoxins in corn by dispersive solid-phase extraction coupled with ultra-performance liquid chromatography with tandem quadrupole time-of-flight mass spectrometry. J. Integr. Agric. 2016, 15, 1656–1666.
- Xing, Y.; Meng, W.; Sun, W.; Li, D.; Yu, Z.; Tong, L.; Zhao, Y. Simultaneous qualitative and quantitative analysis of 21 mycotoxins in Radix Paeoniae Alba by ultra-high performance liquid chromatography quadrupole linear ion trap mass spectrometry and QuEChERS for sample preparation. J. Chromatogr. B 2016, 1031, 202–213.
- Zwickel, T.; Klaffke, H.; Richards, K.; Rychlik, M. Development of a high performance liquid chromatography tandem mass spectrometry based analysis for the simultaneous quantification of various Alternaria toxins in wine, vegetable juices and fruit juices. J. Chromatogr. A 2016, 1455, 74–85.
- Technical report CEN/TR 16059:2010; Food Analysis–Performance Criteria for Single Laboratory Validated Methods of Analysis for the Determination of Mycotoxins. Technical Committee CEN/TC 275/WG 5; European Committee for Standardization: Brussels, Belgium, 2010.
- Al-Taher, F.; Cappozzo, J.; Zweigenbaum, J.; Lee, H.J.; Jackson, L.; Ryu, D. Detection and quantitation of mycotoxins in infant cereals in the U.S. market by LC-MS/MS using a stable isotope dilution assay. Food Control 2017, 72, 27–35.
- Annunziata, L.; Stramenga, A.; Visciano, P.; Schirone, M.; De Colli, L.; Colagrande, M.N.; Campana, G.; Scortichini, G. Simultaneous determination of aflatoxins, T-2 and HT-2 toxins, and fumonisins in cereal-derived products by QuEChERS extraction coupled with LC-MS/MS. Anal. Bioanal. Chem. 2017, 409, 5143–5155.
- Eom, T.; Cho, H.-D.; Kim, J.; Park, M.; An, J.; Kim, M.; Kim, S.-H.; Han, S.B. Multiclass mycotoxin analysis in edible oils using a simple solvent extraction method and liquid chromatography with tandem mass spectrometry. Food Addit. Contam. Part A 2017, 34, 2011–2022.
- European Commission. Commission Regulation (EC) No. 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, L70, 12–34.
- Flores-Flores, M.E.; González-Peñas, E. An LC–MS/MS method for multi-mycotoxin quantification in cow milk. Food Chem. 2017, 218, 378–385.
- Kim, D.-H.; Hong, S.-Y.; Kang, J.W.; Cho, S.M.; Lee, K.R.; An, T.K.; Lee, C.; Chung, S.H. Simultaneous Determination of Multi-Mycotoxins in Cereal Grains Collected from South Korea by LC/MS/MS. Toxins 2017, 9, 106.
- Arroyo-Manzanares, N.; De Ruyck, K.; Uka, V.; Gámiz-Gracia, L.; García-Campaña, A.M.; De Saeger, S.; Di Mavungu, J.D. In-house validation of a rapid and efficient procedure for simultaneous determination of ergot alkaloids and other mycotoxins in wheat and maize. Anal. Bioanal. Chem. 2018, 410, 5567–5581.
- De Boevre, M.; Van Poucke, C.; Ediage, E.N.; Vanderputten, D.; Van Landschoot, A.; De Saeger, S. Ultra-High-Performance Supercritical Fluid Chromatography as a Separation Tool for Fusarium Mycotoxins and Their Modified Forms. J. AOAC Int. 2018, 101, 627–632.
- Du, L.-J.; Chu, C.; Warner, E.; Wang, Q.-Y.; Hu, Y.-H.; Chai, K.-J.; Cao, J.; Peng, L.-Q.; Chen, Y.-B.; Yang, J.; et al. Rapid microwave-assisted dispersive micro-solid phase extraction of mycotoxins in food using zirconia nanoparticles. J. Chromatogr. A 2018, 1561, 1–12.
- Li, X.; Liu, B.; Wang, F.; Ma, X.; Li, Z.; Guo, D.; Wang, Y.; Wan, F.; Deng, L.; Zhang, S. Determination of 16 Mycotoxins in Maize by Ultrahigh-Performance Liquid Chromatography–Tandem Mass Spectrometry. Anal. Lett. 2017, 51, 702–716.
- Meerpoel, C.; Vidal, A.; di Mavungu, J.D.; Huybrechts, B.; Tangni, E.K.; Devreese, M.; Croubels, S.; De Saeger, S. Development and validation of an LC–MS/MS method for the simultaneous determination of citrinin and ochratoxin a in a variety of feed and foodstuffs. J. Chromatogr. A 2018, 1580, 100–109.
- European Commission. Commission Decision of 12 August 2002 implementing Council Directive No. 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, L221, 8–36.
- Solfrizzo, M.; Gambacorta, L.; Bibi, R.; Ciriaci, M.; Paoloni, A.; Pecorelli, I. Multimycotoxin Analysis by LC-MS/MS in Cereal Food and Feed: Comparison of Different Approaches for Extraction, Purification, and Calibration. J. AOAC Int. 2018, 101, 647–657.
- Alcántara-Durán, J.; Moreno-González, D.; García-Reyes, J.F.; Molina-Díaz, A. Use of a modified QuEChERS method for the determination of mycotoxin residues in edible nuts by nano flow liquid chromatography high resolution mass spectrometry. Food Chem. 2018, 279, 144–149.
- SANTE. European Commission. European Commission Health & Consumer Protection Directorate-General. Document No. SANTE 12089/2016. Guidance Document on Identification of Mycotoxins in Food and Feed. Implemented by 01/01/2017. 2016. Available online: https://ec.europa.eu/food/document/download/f16cac78-9318-4f1f-b2fa-efb25d2f1880_en.pdf (accessed on 5 April 2022).
- Dong, H.; Xian, Y.; Xiao, K.; Wu, Y.; Zhu, L.; He, J. Development and comparison of single-step solid phase extraction and QuEChERS clean-up for the analysis of 7 mycotoxins in fruits and vegetables during storage by UHPLC-MS/MS. Food Chem. 2018, 274, 471–479.
- Scarpino, V.; Reyneri, A.; Blandino, M. Development and Comparison of Two Multiresidue Methods for the Determination of 17 Aspergillus and Fusarium Mycotoxins in Cereals Using HPLC-ESI-TQ-MS/MS. Front. Microbiol. 2019, 10, 361.
- Woo, S.Y.; Ryu, S.Y.; Tian, F.; Lee, S.Y.; Park, S.B.; Chun, H.S. Simultaneous Determination of Twenty Mycotoxins in the Korean Soybean Paste Doenjang by LC-MS/MS with Immunoaffinity Cleanup. Toxins 2019, 11, 594.
- De Girolamo, A.; Ciasca, B.; Pascale, M.; Lattanzio, V.M. Determination of Zearalenone and Trichothecenes, Including Deoxynivalenol and Its Acetylated Derivatives, Nivalenol, T-2 and HT-2 Toxins, in Wheat and Wheat Products by LC-MS/MS: A Collaborative Study. Toxins 2020, 12, 786.
- AOAC International. Appendix D: Guidelines for Collaborative Study Procedures to Validate Characteristics of a Method of Analysis. 2005. Available online: http://www.eoma.aoac.org/app_d.pdf (accessed on 5 April 2022).
- EN 17280:2019 Foodstuffs; Determination of Zearalenone and Trichothecenes Including Deoxynivalenol and Its Acetylated Derivatives (3-Acetyl-Deoxynivalenol and 15-Acetyl-Deoxynivalenol), Nivalenol T-2 Toxin and HT-2 Toxin in Cereals and Cereal Products by LC-MS/MS. European Committee for Standardization: Brussels, Belgium, 2019.
- Deng, Y.; Wang, Y.; Deng, Q.; Sun, L.; Wang, R.; Wang, X.; Liao, J.; Gooneratne, R. Simultaneous Quantification of Aflatoxin B1, T-2 Toxin, Ochratoxin A and Deoxynivalenol in Dried Seafood Products by LC-MS/MS. Toxins 2020, 12, 488.
- Gbashi, S.; Njobeh, P.B.; De Saeger, S.; De Boevre, M.; Madala, N.E. Development, chemometric-assisted optimization and in-house validation of a modified pressurized hot water extraction methodology for multi-mycotoxins in maize. Food Chem. 2019, 307, 125526.
- Lago, L.O.; Nievierowski, T.H.; Mallmann, L.P.; Rodrigues, E.; Welke, J.E. QuEChERS-LC-QTOFMS for the simultaneous determination of legislated and emerging mycotoxins in malted barley and beer using matrix-matched calibration as a solution to the commercial unavailability of internal standards for some mycotoxins. Food Chem. 2020, 345, 128744.
- ICH. Harmonised Tripartite Guideline. Guidelines on Validation of Analytical Procedures, Text and Methodol-ogy Q2 (R1). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use (ICH). November 2005. Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed on 5 April 2022).
- EURACHEM. Eurachem Guideline—The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics; EURACHEM: Lisboa, Portugal, 2014; ISBN 978-91-87461-59-0.
- Salim, S.; Sukor, R.; Ismail, M.; Selamat, J. Dispersive Liquid–Liquid Microextraction (DLLME) and LC-MS/MS Analysis for Multi-Mycotoxin in Rice Bran: Method Development, Optimization and Validation. Toxins 2021, 13, 280.
- Zhao, D.-T.; Gao, Y.-J.; Zhang, W.-J.; Bi, T.-C.; Wang, X.; Ma, C.-X.; Rong, R. Development a multi-immunoaffinity column LC-MS-MS method for comprehensive investigation of mycotoxins contamination and co-occurrence in traditional Chinese medicinal materials. J. Chromatogr. B 2021, 1178, 122730.