1. Cell Proliferation
MITF has been shown to enhance or to inhibit proliferation. On one hand, MITF may behave as a melanocyte specific oncogene. MITF is expressed in about 80% of human melanomas (since it is not frequently expressed in desmoplastic melanomas) [
93,
94]; it is amplified in 10% of primary and 20% of metastatic cutaneous melanomas and its expression correlates with decreased 5-year overall patient survival [
95]. Furthermore, a rare germline variant in the
MITF gene (E318K variant) has been linked to a high total nevus count and an increased risk of cutaneous melanoma [
96,
97]. Another illustration of MITF’s role in proliferation is its ability to control the expression of the cell cycle regulators. MITF is known to regulate transcription and expression of the cyclin dependent kinases
CDK2 (cyclin dependent kinase 2) and
CDK4 (cyclin dependent kinase 4) [
95,
98], of
TBX2 (T-box transcription factor 2, a transcription factor of the T-box family), that in turn blocks senescence through repression of p21 and p19 [
99,
100,
101,
102], and of several genes involved in mitosis [
103]. Moreover, MITF exerts a positive control over cell cycle progression through degradation of the growth inhibitor p27 on the one hand, while at the same time it inhibits invasiveness. Both of these functions are carried out through the
DIAPH1 (diaphanous related formin 1) gene [
104]. MITF also drives expression of a large subset of genes involved in lysosome biogenesis and functioning [
81], which triggers increased activity of the lysosome-bound mTORC1 (mTOR complex 1) and global protein synthesis [
105]. Likewise, MITF controls expression of the metabolic factor
PGC1a (PPARG coactivator 1 alpha) [
106,
107]. Enhanced levels of protein synthesis and metabolic activities could allow cancer cells to cope with the metabolic demand related to the high proliferative rate associated with MITF. Consequently, MITF knockdown in human cutaneous melanoma cell lines promotes a growth arrest through induction of a senescence-like phenotype [
108]. On the other hand, MITF has been reported to exert an antiproliferative effect in cutaneous melanoma cells, essentially via p21 regulation [
63].
2. Cell Survival
MITF has been shown to promote cell survival in melanoma cells through several mechanisms. MITF binds E-boxes on the promoters of anti-apoptotic target genes
BCL2, BCL2A1 (BCL2 related protein A1) [
109] and
BIRC7 (baculoviral IAP repeat containing 7) [
31,
61] and participates in transactivation of the receptor tyrosine kinase
MET, thereby increasing the anti-apoptotic effect of the MET ligand HGF (hepatocyte growth factor) [
110]. Under oxidative stress conditions, MITF activates APE1/Ref1 (apurinic-apyrimidinic endonuclease 1 ⁄redox factor-1), which is a protein involved in DNA repair and in redox regulation [
111]. HIF1α is one of the targets of APE1/Ref1 [
112]. HIF1α is also a direct target of MITF. Aligned with this, enhanced
HIF1α expression impaired staurosporin-induced cell death in cutaneous melanoma cells [
113].
3. Epithelial-Mesenchymal Transition and Motile Ability
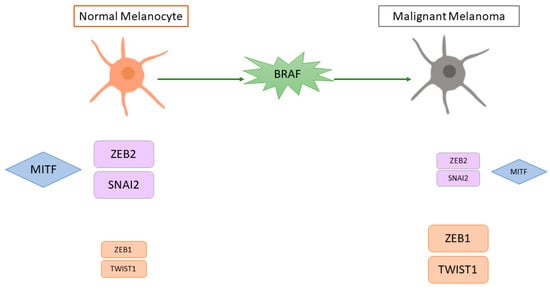
EMT is a complex process in which epithelial cells acquire the characteristics of invasive mesenchymal cells. Melanoma tumour progression and metastasis formation involves a pseudo-EMT process (given the non-epithelial nature of melanoma cells) in which MITF is also involved. Normal cutaneous melanocytes have a high expression of SNAI2 and ZEB2 and a low expression of ZEB1 and TWIST1, while malignant cutaneous melanomas have low SNAI2 and ZEB2 and high ZEB1 and TWIST1 [
114]. Survival analysis in cutaneous melanoma patients showed that high TWIST1 and ZEB1 expression was associated with a shorter metastasis-free survival. Moreover, in vitro BRAF activation caused a switch from a ZEB2high/SNAI2high/ZEB1low/TWIST1low state (similar to normal melanocytes) to a ZEB2low/SNAI2low/ZEB1high/TWIST1high state and MEK inhibitors reversed this switch [
114]. Gene expression profiling revealed that cell lines with high
ZEB1 and
TWIST1 had a de-differentiated gene signature characterised by an upregulation of invasion-associated and TGFβ-regulated genes and downregulation of
MITF and its target genes [
114]. The role of ZEB2 was confirmed in another study, in which human primary cutaneous melanoma samples with high nuclear ZEB2 staining were associated with a better prognosis than tumours with low nuclear ZEB2 staining [
72].
ZEB2 knockdown in mouse melanoma cell lines led to a decrease in
MITF and its target genes and an increase in
ZEB1, thereby leading to a more invasive phenotype. A subsequent study by the same group confirmed that ZEB2 is associated with a proliferative gene signature that includes MITF, while ZEB1 is associated with an invasive gene signature [
115]. A schematic representation of the role of MITF in pseudo-EMT is presented in
Figure 4.
Figure 4. Schematic representation of the putative effect of BRAF mutation on pseudo-epithelial-mesenchymal transition and MITF in cutaneous melanoma. MITF = microphthalmia-associated transcription factor, SNAI2 = snail family transcriptional repressor 2, ZEB2 = zinc finger E-box binding homeobox 2, ZEB1 = zinc finger E-box binding homeobox 1, TWIST1 = twist family BHLH transcription factor 1, BRAF = B-Raf proto-oncogene, serine/threonine kinase. The size of the boxes represents the level of expression (higher expression = bigger box; lower expression = smaller box).
In conclusion, MITF’s role in melanoma cells is important and complex. The rheostat model proposed by the group of Goding provides explanation to the apparent paradox that MITF controls or represses the proliferation or the motile ability of melanoma cells [
104]. MITF expression levels are important but by far not the only criterion in physiology and pathology of melanocytes and melanoma cells. MITF activity also depends on its post-translational modifications (phosphorylation, SUMOylation, ubiquitination) and co-factors (such as p300, BRG1, β-catenin). Hence, cells with low MITF levels are poorly proliferative (likely due to increased p27 expression), dedifferentiated, more mesenchymal and motile [
104], whereas cells with high MITF activity are differentiated and growth-arrested in part through p21 induction [
63]. Supporting this model, positive MITF staining in the primary tumour was associated with a better survival and lower rates of lymph node involvement in cutaneous melanoma patients [
116,
117].
4. Regulation of MITF in Cutaneous Melanoma
Multiple factors and stimuli have been shown to control MITF-M expression in cutaneous melanoma cells. As previously described for skin melanocytes, CREB, PAX3, SOX10, and the Wnt/β-catenin module are also well known upstream regulators of MITF in cutaneous melanoma cells [
19]. In contrast, ATF4 (activating transcription factor 4), and JUN, which both integrate stress signals, repress
MITF expression and trigger cutaneous melanoma cell dedifferentiation [
118,
119,
120]. Another gene that is able to repress MITF is
BRN2, which mediates melanoma cell invasion [
121]. This gene is negatively regulated at the post-transcriptional level by miR-211, which is in turn upregulated by MITF. miR-211 is not the only miRNA involved in the regulation of the invasiveness of cutaneous melanoma cells [
122]. Data regarding miR-182 have shown conflicting results, with some authors reporting it to be upregulated in advanced melanoma and other authors stating that it is downregulated in cutaneous melanoma samples [
123]. Changes in the tumour microenvironment, such as hypoxia or nutrient starvation, may also cause a decrease in MITF expression and greater invasiveness [
92,
118,
124]. Cutaneous melanoma cells exposed to hypoxia have a higher HIF1α expression and lower MITF expression and give rise to larger tumours and more frequent metastases [
92]. The downregulation of MITF caused by hypoxia is dependent on HIF1α, which through the transcription factor Bhlhb2 represses the MITF promoter [
92,
125]. Melanogenesis has been shown to generate an immunosuppressive and mutagenic environment and alters the glycolytic metabolism through HIF1α induction [
126,
127]. Likewise, glutamine starvation reduces MITF level through ATF4 induction [
118]. Additional cues such as TNFα (tumour necrosis factor α) and TGFβ are likely factors that induce the phenotype switch in cutaneous melanoma cells. TNFα can also activate ATF4 and JUN, resulting in dedifferentiated cutaneous melanoma cells [
118,
119,
120]. TGFβ antagonizes MITF function, represses pigmentation and stimulates the motile ability of cutaneous melanoma cells [
128,
129].
5. Clinical Relevance
Both immune checkpoint inhibitors (ICI) and molecularly targeted therapy with BRAF and MEK inhibitors (BRAF/MEKi) are standard options for patients with
BRAFV600-mutated, unresectable, or metastatic melanoma. Options in BRAF wild-type melanoma are limited to ICIs. Despite the progress brought by these treatments, about 50% of the patients reach a therapeutic dead end due to primary or secondary resistance. As mentioned above, cutaneous melanomas are heterogeneous tumours comprised of cells with distinct transcriptomic signatures driving specific behaviours. The two most studied types of melanoma cells are those with a proliferative or invasive phenotype. Due to their high intrinsic plasticity, cutaneous melanoma cells can switch back and forth between these two phenotypes. This plasticity is thought to create intratumour heterogeneity which plays a key role in treatment failure and relapse. Thus, it is of paramount importance to better understand the features of these cell subpopulations to improve treatment efficiency. MITF is thought to be the very determinant of melanoma cell plasticity [
89,
130]. Genomic amplification of the MITF target,
BCL2A1, has been implicated in resistance to BRAF inhibitors by Haq et al. [
109]. Moreover, macrophage-derived TNFα, through increased MITF expression, provides resistance to MAPK pathway inhibitors [
131]. Consequently, inhibition of MITF, by introduction of a dominant-negative MITF mutant in melanoma cells with MITF amplification, or by blocking the TNFα signalling with IκB kinase inhibitors, increased susceptibility of cutaneous melanoma cells to chemotherapeutic agents or MAPK pathway inhibitors, respectively [
95,
131]. These observations suggest that MITF inhibition may represent an option to increase therapy efficacy. However, low MITF expression in cutaneous melanoma cells has also been linked to phenotype switching and drug resistance. Indeed, MITF knockdown in melanoma cells leads to a senescent-like phenotype that is associated with NFkB (nuclear factor kappa-light-chain-enhancer of activated B cell) activation and production of an inflammatory secretome (CCL2, IL6, IL1). This secretome favours a more mesenchymal phenotype, thereby favouring melanoma progression and metastatic dissemination [
132,
133]. Aligned with these findings, Konieczkowski et al. showed that BRAF-mutated melanoma cells with intrinsic resistance to MAPK inhibitors display a low MITF and high NFkB expression [
134]. Likewise, NFkB induction by TNFα led to a decrease in MITF expression and conferred resistance to MAPK inhibitors [
134]. MITF low cells are also associated with an increase in the expression of stem cell genes and reprogramming to a more invasive cell state. Interestingly, MITF activity has been recently shown to be regulated by a direct interaction with RAF proteins in melanoma cells [
135]. By triggering a partial relocation of MITF in the cytoplasm, this interaction might reduce nuclear concentrations of MITF, thereby impacting phenotype switching and therapy efficacy. Melanisation level can also affect the therapy and the clinical outcome of advanced pigmented melanomas [
136,
137,
138].
As highlighted in a review by Ballotti et al., in addition to resistance to targeted therapy, downregulation of
MITF may be also responsible for resistance to immunotherapy [
139] and subsequent melanoma progression [
139]. Inflammatory signals produced by low MITF melanoma cells or by the microenvironment can induce de-differentiation and the consequent loss of melanocyte-specific surface antigens [
139,
140]. Collectively, these observations are in agreement with Müller et al. reporting the existence of two types of resistant cell lines: one with high or normal levels of MITF and one with low MITF level [
141]. Nevertheless, they showed that the cell lines with low MITF levels are more resistant to a wider panel and higher concentrations of MAPK pathway inhibitors than the ones with high MITF [
141].
6. MITF in uveal melanoma
Uveal melanoma is the most common primary intraocular malignancy in adults. Despite originating from melanocytes as well, it is very different from cutaneous melanoma: it uveal melanoma does not have a UV signature, does not carry the typical cutaneous melanoma mutations and does not respond well to target or immunotherapy. A UM usually carries either a GNAQ or a GNA11 mutation [171,172]. When a second mutation occurs, it usually involves one of three genes: BAP1, EIF1AX, or SF3B1 [8,173]. A few studies analysed the presence and function of MITF in UM and, as for cutaneous melanoma, its role seems to be complex: some studies classify MITF as prooncogenic, and others mention its expression as a feature of low-risk tumours. Mouriaux et al. found a positive correlation between MITF staining and proliferative activity [175]. Hippo-YAP/TAZ also emerged as an important signalling pathway downstream of GNAQ/11 that controls UM cell proliferation. Downstream of this module lies PAX3, which controls MITF expression [176]. As such, YAP inhibition suppressed the growth of UM [177,178]. In contrast, MITF has been shown to increase p16 expression in UM, where CDKN2A mutations have rarely been described, supporting the idea that MITF can induce cell cycle arrest and behave as a tumour suppressor gene [64]. miRNAs and epigenetic mechanisms are involved in MITF regulation in UM cells as well: miR-137 through downregulation of MITF, MET, and CDK6 (cyclin dependent kinase 6) and miR-182 through inhibition of MITF, MET, BCL2, and cyclin D2, causing G1 cell cycle arrest, thereby reducing the number of metabolically-active UM cells [179,180]. Increase in miR-137 by the DNA hypomethylating agent 5-aza-20-deoxycytidine
or the histone deacetylase inhibitor trichostatin A (TSA) also represents a therapeutic opportunity to impair UM cell proliferation through MITF inhibition [179]. However, as in cutaneous melanoma, MITF inhibition might be associated with a stem cell-like phenotype. Matatall et al. showed that BAP1 knockdown was associated with an increase in expression of stem cell markers (NANOG), and loss of pigmentation markers (MITF, TYR, and DCT) and with the capacity of UM cells to form anchorage-independent colonies. These data suggest that BAP1 loss induced a dedifferentiated and a stem-like phenotype in UM cells, although this remains to be fully demonstrated [181].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23116001