Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Microglia represent 10–15% of the total cells in the brain. These cells are considered as the resident macrophages and the first defense line in the central nervous system (CNS) against pathogens [15]. In physiological conditions, microglia have a high capacity to respond to changes in the CNS microenvironment owing to their processes.
- neuroprotection
- microglia
- Brain
- Ischemia
- pMCAo
1. Microglia
Microglia represent 10–15% of the total cells in the brain. These cells are considered as the resident macrophages and the first defense line in the central nervous system (CNS) against pathogens [15]. In physiological conditions, microglia have a high capacity to respond to changes in the CNS microenvironment owing to their processes [16]. These changes may be triggered by microorganisms such as bacteria or viruses, but also by neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, or cerebral ischemia. Particularly, microglia activation is one of the first events that occurs after an insult such as brain ischemia [17], taking place from minutes to a few hours after the start of the episode [18] (Figure 2).
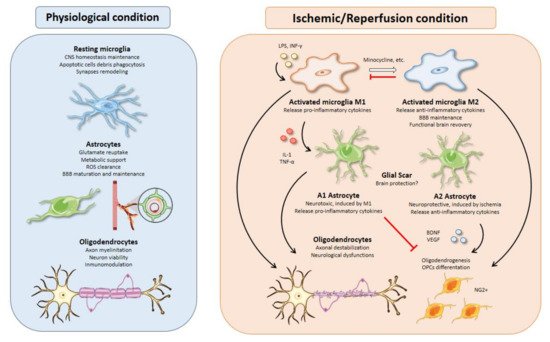
Figure 2. Glial cell functions, responses, and interactions in physiological and I/R conditions. Microglial activation is one of the earliest events after brain ischemia. The inflammatory landscape generated in the ischemic brain by inflammatory cytokines, debris, or molecules released from dead cells triggers microglia activation. Activated microglia are typically divided into M1 and M2 phenotypes. M1 microglia present a pro-inflammatory profile releasing cytokines such as IL-1 or TNF-α that can polarize astrocytes toward a neurotoxic phenotype, aggravating the inflammatory response. On the other hand, M2-microglia release anti-inflammatory cytokines, sustain blood brain barrier (BBB) integrity, and stimulate oligodendrogenesis through oligodendrocyte progenitor cells’ (OPCs) differentiation into NG2+ cells, thus promoting functional recovery after brain ischemia. All therapeutic strategies based on microglia are focused on minimizing the effects of M1-microglia using drugs like minocycline to switch phenotype from M1 to M2. Besides, M2 microglia could promote an inhibition of M1 microglia. Activated astrocytes are usually classified into A1 and A2 phenotypes. In line with M1 microglia, A1 astrocytes present a neurotoxic profile releasing pro-inflammatory cytokines with inhibitory effects over oligodendrogenesis and OPCs’ differentiation. In contrast, A2 astrocytes have neuroprotective functions releasing anti-inflammatory cytokines and trophic factors, such as BDNF or VEGF with similar effects over oligodendrocytes as those of M2 microglia. Oligodendrocytes are especially sensitive to oxidative stress and excitotoxicity generated during brain ischemia. Demyelination affects neurons owing to axonal destabilization, generating neurological dysfunctions. Moreover, these events are aggravated in the presence of M1 microglia and A1 astrocytes. At the same time, trophic factors released by M2 microglia and A2 astrocytes increase oligodendrogenesis and OPCs’ differentiation in order to repair damaged white matter in the injury area.
2. Microglial Activation Timing upon Brain Ischemia
Morioka et al. [19], using Nissl staining, described an activation of microglia after 24 h of permanent middle cerebral artery occlusion (pMCAO) in cortical and thalamic regions. Years later, Schoroeter et al. [20] showed that, after 24–72 h of pMCAO, microglia presented star shape with thick and short processes located near the damaged area, while 6 days after pMCAO, microglia acquired an amoeboid shape in the same area. Thanks to the development of imaging techniques such as MRI, there has been deep progress in the study of microglia activation after cerebral ischemia. Rupalla et al. [21] described the activation of microglia as early as 30 min after pMCAO, showing hypertrophic cell body and processes in the penumbra. This revealed a high capacity of microglia to activate after a disruption in tissue homeostasis. Recently, a series of studies revealed the presence of activated microglia in the acute phase [22], sub-acute phase [23], and chronic phase [24] of IS. The aforementioned morphology changes are associated with different activated microglia functions. Some of these functions include an increase in phagocytosis rate, release of anti- or pro-inflammatory cytokines, proliferation, and migration [25].
3. M1 versus M2 Microglial Profiles
Under physiological conditions, microglia are in a “resting” status, from which they are able to lead a wide range of responses upon detection of changes in the environment like modulation of their dynamic processes, removing of debris and apoptotic cells by phagocytosis and remodeling of synapses [26]. Resting microglia present a low expression profile of surface molecular markers that include CD45, MHC-II, CD80, CD86, and CD11cc [27]. After an ischemic insult, activated microglia change this profile, presenting high expression of CD45, MHC-II, or CD86, among others. Moreover, Iba1, IB4, F4/80, and CD68 can also be used to identify activated microglia in this context [28]. Interestingly, there are differences in activated microglia molecular profile between the penumbra and ischemic core that could be used to delimit both regions. For example, the activated microglia in the penumbra are MHC-II+, associated with anterograde degeneration, while the core’s activated microglia are MHC-I+ phagocytic cells [29]. Activated microglia have been classically categorized into two general groups according to the paradigm of macrophage activation (Figure 2). In general, the scientific community named a pro-inflammatory profile as M1 activated microglia, while an anti-inflammatory profile was termed M2 activated microglia [30,31]. Despite the persistence of this binary classification, there are many works supporting a heterogeneity in the population of activated microglia and a coexistence of intermediate phenotypes as M2-a, M2-b, or M2-c [32,33,34]. Precisely identifying the different subpopulations of activated microglia that appear after an ischemic insult could be of great relevance for the development of effective treatments. Kanazawa et al. [35] defined a temporal polarization after ischemic stroke by activated microglia markers. They described a majority of M2 activated microglia population during the first 24 h after brain ischemia followed by an increase in M1 microglia population. Other studies support the idea that the balance between both activated phenotypes could determine the neurodegenerative disease progression, so that a majority of the M1 population is associated with a worse clinical prognosis [36]. Interestingly, activated microglia are known to switch from one profile to another and a great number of therapies are focused on this property [37,38,39]. M1 activated microglia contribute to an increase in the inflammation and cytotoxicity levels through the release of the cytokines TNF-α and IFN-γ; interleukins such as IL-1β, IL-6, IL-15, IL-18, and IL-23; chemokines like CCL2 and CXCL10; the metalloproteinases (MMPs) MMP-3 and MMP-9; and reactive oxygen/nitrogen species (ROS/RNS) [40,41]. On the other hand, M2 activated microglia promote the recovery of injured tissue and decrease inflammatory levels by secreting molecules such as IL-4, IL-10, IL-13, TGF-β, IGF-1, the neurotrophic factor BDNF, and vasoactive proteins [42].
4. Inductors of Microglial Activation in Stroke
There are several pathways that lead to microglial activation after brain ischemia. Classic activation is associated with M1 activated profile, while alternative activation is associated with the M2 activated profile [43]. There is a great range of molecular mechanisms underlying microglia activation after cerebral ischemia. Damage-associated molecular patterns (DAMPs) are molecules released in a passive way from cell debris or apoptotic cells after brain ischemia, driving microglial activation towards pro- or anti-inflammatory phenotypes [44]. Among these molecules, high mobility group box 1 (HMGB1) protein appears in the early stages of stroke and is recognized by several toll-like receptors (TLRs) like TLR2 or TLR4, which trigger an inflammatory response through the release of pro-inflammatory cytokines in an NF-κB-dependent process. In fact, TLR4 blockade has shown to be protective against brain ischemia with a reduction of the infarcted area, which could be due to a decrease in pro-inflammatory cytokines secreted by microglia [45]. Peroxiredoxins (Prdxs) constitute another important group of DAMPs with redox-activity. Prdx-1, Prdx-2, Prdx-5, and Prdx-6 are secreted by necrotic cells in the brain early after stroke inducing the release of pro-inflammatory cytokines, which are then recognized by TLR2 and TLR4 [46].
Initial disturbance of BBB integrity after an ischemic insult is also known to recruit and activate microglia, which start to secrete pro-inflammatory cytokines including IL-1β, TNF-α, and IL-6. IL-1β strongly induces activation of the astrocytes implicated in the neurovascular unit (NVU), promoting disruption of this functional structure, and thus leading to a metabolic uncoupling between neurons and the proximal blood flow [47]. The same population of activated microglia increases paracellular permeability of the surrounding blood vessels and further disrupts the NVU mainly through altered cytoskeletal organization, defective tight junction proteins’ (TJPs) expression, and MMPs’ release [37]. On the other hand, the M2 microglia population has been shown to exert angiogenic functions and promote BBB integrity, mainly through expression of TJPs [48].
Glutamate receptors (mGluRs) are also considered as microglial-related targets in brain ischemia. It is known that blockage or ablation of mGluR5 reduces acute microglial activation and promotes neuroprotection and neurofunctional recovery [49,50]. On the other hand, purinergic receptors, like P2X4R, P2X7R, and P2Y6R, have been related to neuroprotection mediated by microglia after brain ischemia owing to their anti-inflammatory effects [51,52]. This is a very complex process in view of the wide variety of receptors and channels on the microglial cell surface that get activated at the same time, triggering their own signaling cascade, and that must be tightly regulated to guarantee a harmonized response [53].
5. Therapies Based on Activated Microglia
From a therapy point of view, the available data suggest different alternatives to use activated microglia as a therapeutic target for brain ischemia. In vitro experiments using oxygen and glucose deprivation (OGD) offer an excellent model to study activated microglia response. These experiments usually focus on the molecular mechanisms underlying microglial polarization through the use of different agents such as LPS and IFN-γ, as a way to study the possibility to revert detrimental M1 profile and induce a neuroprotective M2 profile (Figure 2). On the other hand, in vivo experiments offer a better approach to study the role of activated microglia after an ischemic insult and their relation to other cell types, which remains poorly understood. By collecting data through these two approaches, we could know more specifically the response of activated microglia in this pathological context and develop effective strategies to reduce the detrimental effects of brain ischemia.
Minocycline is an antibiotic of the tetracycline family and it is the main compound used to abrogate inflammation induced by microglia activation [54]. Minocycline promotes a switch between microglial phenotypes, reducing the expression of M1 profile mRNA levels (IL-1β, IL-6, iNOS, and TNFα) and increasing typical M2 mRNAs (Arg-1, IL-10, TGF-β, and Ym1) [55]. Such an effect results in a reduction of the amoeboid morphology near the ischemic cortex and the infarct area [56]. Minocycline is also known to reduce cell death through the STAT1/STAT6 pathway [55]. Moreover, LPS-activated BV-2 microglial cell line presented a reduction in pro-inflammatory markers (CCL2, IL-6, and iNOS), with a concomitant decrease in caspase 3/7 activity and cell death [57]. Additionally, there are many studies supporting the idea that minocycline also increases the integrity of blood vessels, contributing to maintenance of the BBB integrity by an M2 microglia polarization [58,59].
Minocycline is one of the few glial-related components that have been tested in several clinical trials with promising results. An open-label, evaluator-blinded study of 152 patients showed minocycline, administered within 6 to 24 h of onset of stroke, to be associated with significantly lower National Institutes of Health Stroke Scale score and modified Rankin Score (mRS) compared with placebo [60]. By contrast, a multicenter randomized, double-blind, placebo controlled trial, “Neuroprotection With Minocycline Therapy for Acute Stroke Recovery Trial” (NeuMAST), in which patients were orally administered with either minocycline or placebo within 3 to 48 h of symptom onset, failed to prove any long-term beneficial effects over neurological outcome [61].
Apart from minocycline, there are a number of other compounds with promising efficiency in reverting microglia-derived detrimental effects in IS. Melatonin administration post-stroke is able to switch from M1 to M2 activated microglia through the STAT3 pathway, increasing secretion of anti-inflammatory cytokines and, therefore, reducing the damaged brain area [62]. Using GAPIs (a molecular cocktail from Ginkgo biloba), Zhou et al. [63] showed a reduction in the secretion of pro-inflammatory cytokines in BV-2 microglial cells, which acquired an M2 phenotype. Protocatechuic acid used after a cerebrovascular accident reverts M1-commited microglial cells and promotes expression of M2 markers [64]. Mechanistic target of rapamycin (mTOR) inhibitors like rapamycin are known to reduce the inflammatory microenvironment and infarct volume, decreasing the number of Iba1+ cells and the expression of M1 markers after pMCAO [65]. ABIN1, a NF-κB inhibitor, has also been shown to attenuate both microglia activation and the levels of pro-inflammatory cytokines after brain ischemia [66]. Interestingly, L-3-n-butylphthalide, an extract from seeds of Apium graveolens Linn, administered during 7 consecutive days after 45 min of pMCAO, improved the sensorimotor functions and reduced brain infarct volume, leading to an M2 microglia polarization [67].
Electroacupuncture (EA) is another neuroprotective strategy known to inhibit inflammation after brain ischemia, with many physical points available to apply this therapy including Baihui (GV20), Shuigou (GV26), Neiguan (PC6), Hegu (LI4), and Taichong (LR3), among others. There are some studies reporting the use of EA preconditioning to decrease pro-inflammatory effects triggered by brain ischemia. Using EA pre-treatment before pMCAO in rats, Liu et al. [68] showed a significant reduction of the infarct volume paralleled by functional motor recovery. From a molecular point of view, there is a reduction in the levels of different pro-inflammatory effectors such as TNF-α, IL-1β, and IL-6 in the damaged area and blood serum after I/R. Moreover, EA blocks the nuclear translocation of NF-κB (p65), preventing the expression of p38 MAPK and MyD88. In addition, another work explained that EA was able to induce a α7nAChR-dependent significant decrease of infarcted area and improve the neurological outcome [69].
This entry is adapted from the peer-reviewed paper 10.3390/cells10071639
This entry is offline, you can click here to edit this entry!