Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Chemokines are chemotactic cytokines, whose most important function is the chemoattraction of immune cells. CXC motif chemokine ligand 1 (CXCL1), a CXC chemokine, is also known as growth-regulated (or -related) oncogene-α (Gro-α) and melanoma growth-stimulatory activity (MGSA). The role CXCL1 plays in the physiology of the nervous system is described.
- CXCL1
- Alzheimer’s disease
- Epilepsy
- neurodegenerative diseases
- neuroinflammation
1. Prenatal Development of the Brain
CXCL1 may be important in prenatal brain development. However, the significance of CXCL1 in prenatal development in humans has been poorly studied due to obvious bioethical problems with such research. The importance of ligands for CXCR2 in this aspect has been much better studied in animals.
Experiments on rats show that ligands for CXCR2 are important in axon morphogenesis, a process regulated by hepatocyte growth factor (HGF). For example, this factor in rats increases the expression of cytokine-induced neutrophil chemoattractant-3 (CINC-3), a CXC chemokine important in axon outgrowth and axon branching [1]. Ligands for CXCR2 are known to reduce axon outgrowth of dorsal root ganglia neurons in adult rats [2], thereby reducing peripheral nervous system regeneration. However, the significant differences in ligands for CXCR2 between humans and rats make it impossible to determine which human chemokine is responsible for the aforementioned properties [3].
CXCL1 may be important in the functions of oligodendrocyte progenitors. For example, research on rats has shown that CINC-1 increases the proliferation of these cells, exhibiting synergy with platelet-derived growth factor (PDGF) [4][5]. In humans, this role is played by CXCL1 [6]. Expression of this chemokine occurs in the cortical ventricular/subventricular zones, the sites of proliferation of oligodendrocyte progenitors. CXCL1 can inhibit the migration of oligodendrocyte progenitors, as shown by research on the effects of CINC-1 in the rat brain and spinal cord [5][7], indicating that this chemokine regulates the location of oligodendrocytes.
2. Neurogenesis, Hippocampus and Neural Stem Cells
CXCR2 ligands are important in the hippocampus, particularly for neurogenesis, as demonstrated by experiments in mice and rats. In mice, KC increases hippocampal neurogenesis [8]. The expression of this chemokine occurs in the subgranular zone of the dentate gyrus in the hippocampus [9]. KC induces neural stem cell proliferation but inhibits the differentiation of these cells into astrocytes [10]. At the same time, studies in rats have shown that CINC-1 is subject to expression in damaged parts of the brain, particularly in the damaged striatum, where this chemokine causes the recruitment of progenitor cells from subventricular zone [11], that is, it participates in the regeneration of neural tissue in the brain. Nevertheless, the cited facts are yet to be confirmed in humans.
CXC chemokines can also interfere with hippocampal neurogenesis in some models. In particular, during neuroinflammation [9]. KC in a mouse model causes senescence of hippocampal neuronal progenitor cells. This reduces neurogenesis in this brain structure. These results were confirmed on human hippocampal neuronal progenitor cells and CXCL1 [9]. The observed effect was sex-dependent, as female sex hormones counteract the increase in KC expression by pro-inflammatory factors in the hippocampus [9].
3. Addiction and Reward System
CXCL1 may also play a role in the mechanisms of addiction. In mice, administration of cocaine increases KC expression in the prefrontal cortex, in a process dependent on the dopamine D1 receptor [12]. KC, through the activation of CXCR2, is an important part of reward system activation in the brain when exposed to cocaine. To date, there has been no such research on humans and for this reason it is not known whether CXCL1 or any other ligands for CXCR, is significant in the action of cocaine in the human brain. There are no studies regarding the effect of CXCL1 on the reward system during daily activities [12].
4. Alzheimer’s Disease
Alzheimer’s disease is a neurodegenerative disease that causes dementia [13]. It is estimated that tens of millions of people suffer from this disease worldwide, mostly in advanced age. An important pathomechanism in Alzheimer’s disease is the accumulation of amyloid β (Aβ) in amyloid plaques and tau protein in neurofibrillary tangles, which cause neurodegeneration of the brain. Another component of Alzheimer’s disease is neuroinflammation, including increased CXCL1 expression and action.
In Alzheimer’s disease patients, CXCL1 levels are elevated in cerebrospinal fluid [10][14] and the brain [15]. CXCL1 is produced by neurons in the brain of Alzheimer’s disease patients [15]. In pathological brain tissue, CXCL1 activates the CXCR2 receptor on neurons, which causes the activation of ERK MAPK and glycogen synthase kinase 3β (GSK3β) leading to the hyperphosphorylation of tau (Figure 1) [15][16]. Then, prolonged exposure of neurons to CXCL1 results in Tau cleavage at Asp421 by caspase-3 [16]. Such truncated tau proteins may be the primer of neurofibrillary tangle formation [16]. Hyperphosphorylation also increases the aggregation capacity of tau proteins. CXCR2 activation has been found to increase γ-secretase activity, a protease whose substrate is amyloid precursor protein (APP) [17][18][19]. This causes the release of Aβ which contributes to the formation of amyloid plaques.
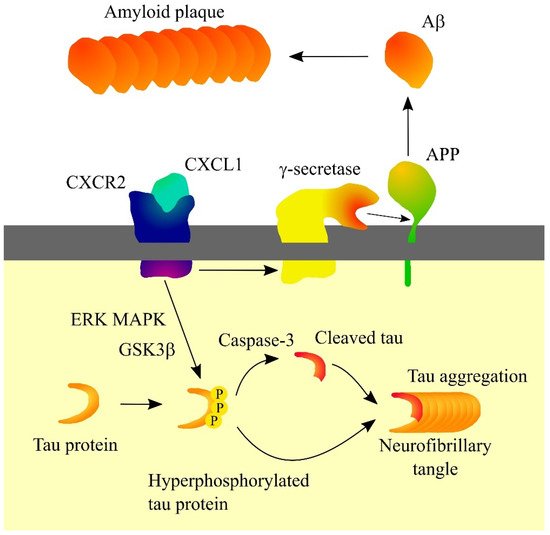
Figure 1. Significance of CXC motif chemokine ligand 1 (CXCL1) in Alzheimer’s disease. CXCL1, through its receptor CXC motif chemokine receptor 2 (CXCR2), causes hyperphosphorylation of tau protein which leads to a proteolytic cleavage of tau protein, which enables the formation of a neurofibrillary tangle. CXCR2 also increases γ-secretase activity. This results in increased release of amyloid β (Aβ) and increased amyloid plaque formation. Abbreviations: Aβ—amyloid β; APP—amyloid precursor protein; CXCL1—CXC motif chemokine ligand 1; CXCR2—CXC motif chemokine receptor 2; ERK—extracellular signal-regulated kinase; GSK3β—glycogen synthase kinase 3β; MAPK—mitogen-activated protein kinase; PPP—hyperphosphorylation; Source: own elaboration.
CXCL1 may also participate in mechanisms that inhibit the progression of Alzheimer’s disease. Patients with this disease have elevated CXCL1 expression in blood monocytes [20]. In addition, expression of CXCR2 on brain microvascular endothelial cells is increased by Aβ [20]. This indicates that the CXCL1→CXCR2 axis is responsible for transendothelial migration of monocytes into the brain. Such monocytes differentiate into bone marrow-derived microglia, involved in the elimination of Aβ plaque deposition, inhibiting the progression of Alzheimer’s disease [20][21].
5. Epilepsy
Epilepsy, a brain disease whose major symptoms include unprovoked seizures, is found in about 80 million people in the world [22]. Ligands for CXCR2 contribute to its pathogenesis—as shown, for example, by increased KC expression in the brain of epileptic mice, particularly in the hippocampus [23]. KC decreases astrocytic glutamate reuptake [24], which causes an increase in glutamate concentration at synapses and leads to seizures. CXCL1 does not seem to have the same role in epilepsy in humans as it does in mice. In contrast to TNF-α and CXCL9, CXCL1 levels are not elevated in the cerebrospinal fluid of patients with epilepsy [25]. In comparison, CXCL8 expression is increased in the brain of patients with epilepsy [23] which suggests its role is similar to that of KC in mouse epilepsy. However, further studies in this direction are required.
6. Herpes Simplex Virus Type 1 (HSV-1) Encephalitis and Herpetic Stromal Keratitis (HSK)
Herpes simplex virus type 1 (HSV-1) is a virus possessing double-stranded DNA of fairly large size: 152 kbp [26]. HSV-1 belongs to the Alphaherpesvirinae subfamily, which also includes HSV-2 and Varicella zoster virus (VZV)/HSV-3. It is estimated that approximately half of the population has had contact with HSV-1 [27]. Infection with this virus occurs when fluids containing HSV-1 come into contact with mucous membranes [26]. Then HSV-1 infects the mucosal epithelium and then sensory neurons near the site of primary infection where HSV-1 progresses to latent infections [26][28].
HSV-1 can also enter the brain, where it can cause quite rare cases of severe encephalitis [28] at 0.25–1.2 per 100,000 population per year [28]. HSV-1 encephalitis is associated with an increase in the expression of pro-inflammatory cytokines in the brain [29]. In particular, experiments on mice have shown an increase in KC expression in astrocytes under the influence of HSV-1 [30]. There is also an increase in KC expression in astrocytes and neurons under the influence of pro-inflammatory cytokines such as IL-1α. KC causes infiltration of the brain by neutrophils which results in blood-brain barrier damage and an excessive immune system response, resulting in brain damage and death. The KC→CXCR2 axis and neutrophils do not affect HSV-1 viral load in the brain as shown by experiments in mice [30]. As monocytes and the CCL2→CCR2 axis are responsible for fighting the virus in the brain, the KC→CXCR2 axis represents a convenient therapeutic target against HSV-1 encephalitis in a mouse model. With mouse KC being a paralog for human CXCL1 [3][31][32], it needs to be confirmed if changes in CXCL1 expression occur in patients with HSV-1 encephalitis, to determine its role in this disease in humans.
HSV-1 has also been found to infect the cornea, leading to herpetic stromal keratitis (HSK) [33], often resulting in blindness. Cornea infection by HSV-1 is associated with an increase in KC expression due to the action of IL-17A, as shown by experiments in a mouse model [34]. This leads to infiltration of the cornea by neutrophils [34]—cells involved in fighting the viral infection but also in damaging the cornea [33]. Among other things, neutrophils secrete vascular endothelial growth factor A (VEGF-A) and enhance the action of this growth factor by secreting MMPs, which leads to corneal neovascularization [34]. Depending on the study, either KC [33][34] or MIP-2 [35][36] plays a major role in neutrophil infiltration into the cornea during HSV-1 infection. Both chemokines are CXC chemokines [3][31] and are paralogs to human CXCL1, so the significance of CXCL1 needs to be confirmed in human patients with the aforementioned diseases.
7. Ischemic Stroke
Ischemic stroke results from brain arterial occlusion followed by reduced blood flow to various parts of the brain [37]. Very often it ends in death or extensive brain damage and disability. It is estimated that nearly 10 million cases of ischemic stroke occur annually, making it one of the most common diseases in the world [37].
CXCL1 expression has been found to be closely associated with ischemic stroke. Therefore, individuals with the T allele of rs3117604, which is located in the CXCL1 promoter, have an increased predisposition to ischemic stroke [38].
Patients with ischemic stroke have elevated levels of CXCL1 in cerebrospinal fluid [39] and at the same time CXCL1 levels are associated with the extent of ischemic stroke. Increased CXCL1 expression has also been shown in brain tissue affected by ischaemic stroke [40]. Nevertheless, depending on the literature cited, CXCL1 levels in the blood in patients with ischemic stroke are either unchanged [39] or lower [41]. With the discussed CXCL1 levels in the blood, this may be related to gender. In women with ischaemic stroke, CXCL1 levels in the blood are lower, while in men they are higher [42].
Expression of KC in the brain at the ischaemic stroke takes place in astrocytes, as shown by experiments on mice (Figure 2) [40]. γδ T cells, which produce IL-17A, are responsible for the induction of KC expression. Expression of KC in astrocytes also depends on TNF-α produced by macrophages [40]. At the same time, CD4+ T cells produce interferon-γ (IFN-γ), which induces TNF-α expression in macrophages.
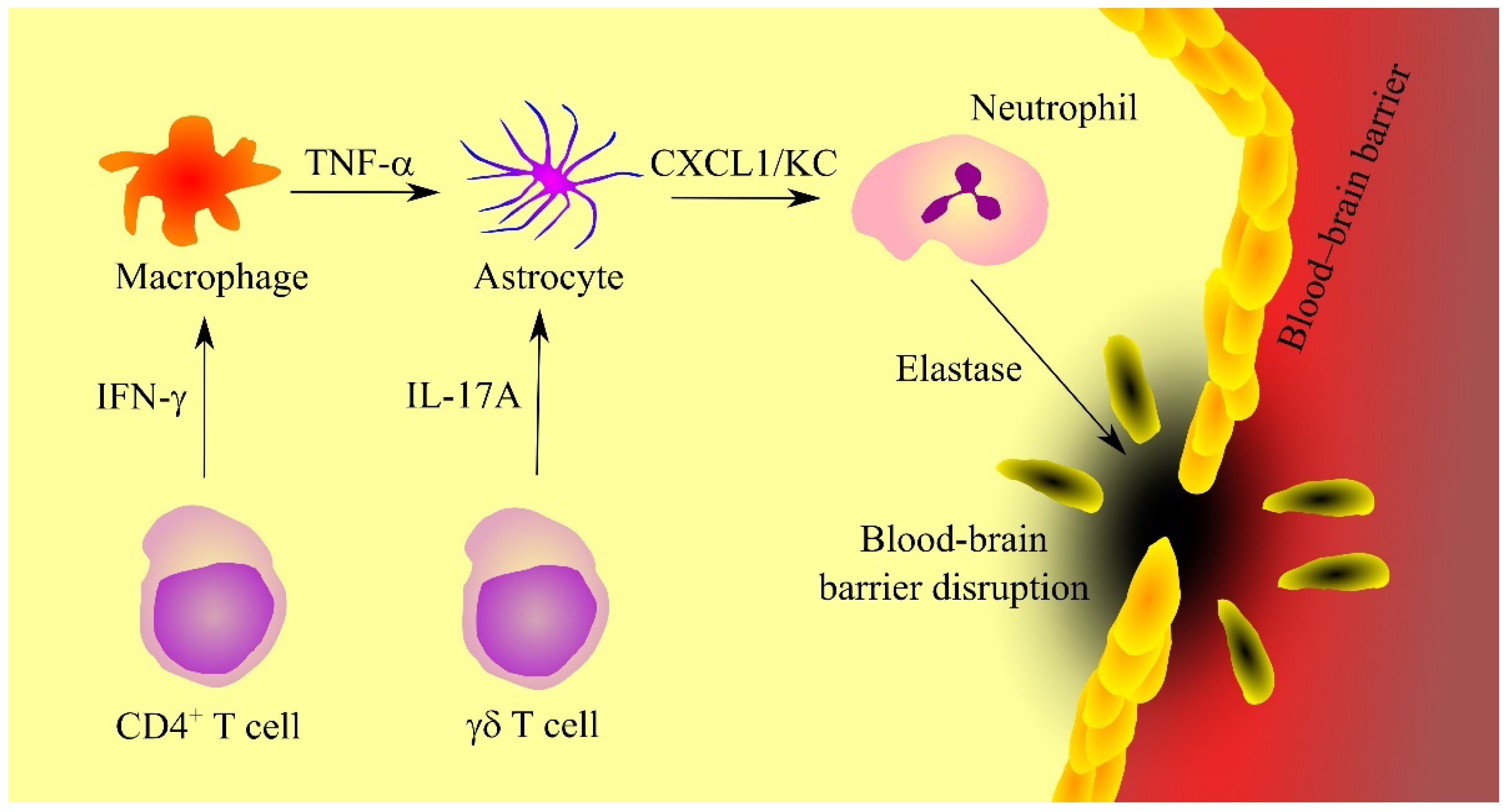
Figure 2. The significance of CXC motif chemokine ligand 1 (CXCL1)/keratinocyte-derived chemokine (KC) in ischemic stroke. Ischaemic stroke is associated with neuroinflammation caused by γδ T cells and CD4+ T cells that respectively secrete interleukin-17A (IL-17A) and interferon-γ (INF-γ). INF-γ increases tumor necrosis factor α (TNF-α) production in macrophages. TNF-α and IL-17A increase the secretion of CXCL1/KC in astrocytes. This chemokine causes recruitment of neutrophils, cells which secrete elastase, an enzyme that causes blood-brain barrier disruption. Abbreviations: CXCL1—CXC motif chemokine ligand 1; IL-17A—interleukin-17A; INF-γ—interferon-γ; KC—keratinocyte-derived chemokine; TNF-α—tumor necrosis factor α; Source: own elaboration.
Expression of ligands for CXCR2 during ischemic stroke may also depend on miRNAs. In rats, an ischemic stroke is associated with promoter hypermethylation of miR-532-5p [43], which reduces the expression of the miRNA that is directly downregulating the expression of the rat paralog for human CXCL1. That means that this downregulation of miR-532-5p expression induces an upregulation of CXCL1 expression. In contrast, during ischemic stroke in mice, there is a downregulation of miR-429 expression in brain microvascular endothelial cells [44]. This miRNA downregulates the expression of KC, the mouse paralog for human CXCL1, which results in increased expression of KC. As a consequence of increased CXCR2 ligand expression, neutrophils infiltrate into brain tissue undergoing ischemic stroke. It is not known whether these cells have a destructive effect on brain tissues in patients with ischemic stroke. Studies in mice have shown that a CXCR2 receptor inhibitor does not affect the level of motor dysfunction after ischemic stroke [45]. However, neutrophils can disrupt the blood-brain barrier (BBB) integrity through elastase secretion [46]. Because the contribution of neutrophils to brain damage after ischemic stroke has not been sufficiently investigated, more thorough studies in this direction are needed.
8. Major Depression
Major depression is a mental illness that affects approximately 6% of the population and its pathogenesis may be related to inflammation [47]. In mice, chronic stress causes activation of the inflammasome in the hippocampus [48][49], increasing the production of the pro-inflammatory cytokines IL-1β and interleukin-18 (IL-18). This leads to inflammatory responses associated with increased KC production [49]. KC, through activation of its receptor CXCR2 and subsequent activation of GSK3β, induces depression-like behaviors.
The mechanism of CXCL1 involvement in major depression in humans appears to differ significantly from the animal model. Depressed suicidal persons experience a reduction in CXCL1 expression in the prefrontal cortex, similar to the reduction in the expression of CXCL2 and CXCL3 [50]. Plasma CXCL1 levels are also reduced in depressed humans [51], particularly in elderly patients [52] and adolescents [53]. One paper shows that in elderly patients, plasma CXCL1 may be slightly increased in depression, although the results were not statistically significant [54]. In contrast, depressed patients have been shown to have much higher levels of CXCL7 and CXCL8 in their blood [55]. It is likely that these chemokines have a function in depression that KC plays in mice, but another mechanism cannot be ruled out.
There are no studies showing correlations between depression on CXCL1 expression in the hippocampus in humans. It is possible that, as in experimental animals, depression is associated with an increase in CXCL1 expression in this brain structure in humans. If, in contrast, the human hippocampus showed a decrease in CXCL1 expression, it would indicate that CXCL1 plays an important role in brain function in humans and a decrease in the expression of this chemokine could cause depression. This would likely be related to hippocampal neurogenesis [8] or impaired oligodendrocyte function and myelination [56][57][58].
9. Multiple Sclerosis
Multiple sclerosis is a neurodegenerative and autoimmune disease of the brain and spinal cord, with an estimated incidence of 0.2 to 240 people per 100,000, depending on the population studied [59]. Myelin-reactive T cells are the main element responsible for the pathogenesis of this disease [60]. Following the action of these cells, demyelination and inflammatory reactions result in the dysfunction of the nervous tissue.
CXCL1 is a significant element in the course of multiple sclerosis. This chemokine has been shown to have both neuroprotective and inhibitory properties in the progression of multiple sclerosis [56], as well as being one of the elements contributing to neurodegeneration [61]. Nevertheless, it seems that the destructive properties of CXCL1 are predominant, as in patients with multiple sclerosis, plasma CXCL1 levels are correlated with clinical disability [62].
CXCL1 expression is increased in the brains of patients with multiple sclerosis [57]. In particular, it is found in areas of demyelination [61][63]. Elevated CXCL1 levels have also been shown in multiple sclerosis patients in blood [62] and cerebrospinal fluid [64][65]. However, these data are debatable as some studies have shown that CXCL1 levels in cerebrospinal fluid were not different in patients with multiple sclerosis [66][67]. This discrepancy may be due to the fact that CXCL1 could be a significant factor in the development of multiple sclerosis in just the early stages of the disease [68][69].
In multiple sclerosis, CXCL1 is produced by activated microglia [70] and astrocytes (Figure 3) [57][58]. Also, CXCL1 expression is dependent on CD4+ T helper type 17 (Th17) cells which produce IL-17 [62][71][72]. This cytokine increases the expression of CXCL1. IL-17 has toxic effects on oligodendrocytes [73]. IL-17 appears to be significant in the early stages of multiple sclerosis [68]. As the disease progresses and treatment is given, the concentration of IL-17 in cerebrospinal fluid decreases. There are also indications that CXCL1 expression and disease progression in patients with multiple sclerosis may also be inhibited by IFN-γ [74]. In mice, IFN-γ inhibits the expression of MIP-2 but not KC [72]. However, as it is very difficult to find the exact counterparts of these two CXC chemokines [3], there is a need for experiments investigating the effect of IFN-γ on CXCL1 expression in human neural tissue during multiple sclerosis. IFN-γ increases the production of nitric oxide (NO) which enhances the ability of neutrophils to inhibit autoreactive T cells [74][75], which inhibits the progression of multiple sclerosis.
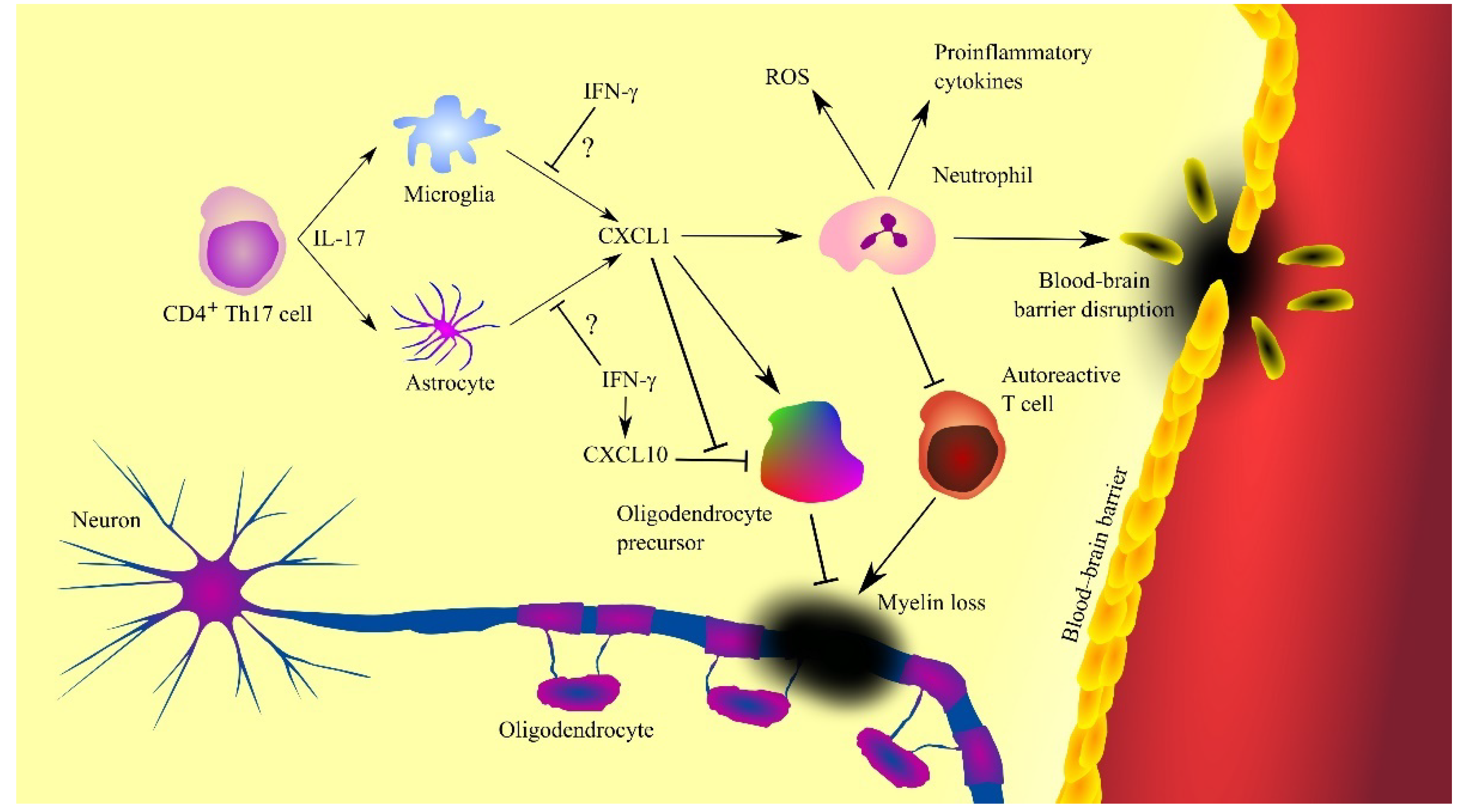
Figure 3. Involvement of CXC motif chemokine ligand 1 (CXCL1) in the mechanisms of multiple sclerosis. CXCL1 expression is increased by interleukin-17 (IL-17) produced by T helper type 17 (Th17) cells, which triggers the recruitment of neutrophils, cells contributing to the progression of the disease by producing reactive oxygen species (ROS), proinflammatory cytokines and causing blood-brain barrier (BBB) disruption. However, neutrophils can also inhibit the destructive effects of autoreactive T cells. CXCL1 also acts on oligodendrocyte precursor cells by inducing their proliferation and inhibiting CXC motif chemokine ligand 10 (CXCL10)-induced apoptosis of these cells. Oligodendrocyte precursor cells differentiate into oligodendrocytes, leading to remyelination and disease regression. Abbreviations: CXCL1—CXC motif chemokine ligand 1; CXCL10—CXC motif chemokine ligand 10; IFN-γ—interferon-γ; IL-17—interleukin-17; ROS—reactive oxygen species; Th17—T helper type 17 cells; Source: own elaboration.
Inflammation has a destructive effect on oligodendrocyte progenitor cells as shown by experiments on human embryonic stem cell-derived oligodendrocyte progenitor cells [76] and murine oligodendrocyte progenitor cells [77]. This is related to the induction of apoptosis of these cells by CXCL10/γ interferon inducible protein 10 (IP-10), a chemokine whose expression is induced by IFN-γ [76][77]. In contrast, CXCL1 and thus CXCR2 receptor activation inhibits CXCL10/IP-10-dependent apoptosis of oligodendrocyte progenitor cells [76][77]. IFN-γ and CXCL10/IP-10 are elevated in the cerebrospinal fluid of patients with multiple sclerosis [65]. Therefore, the adverse effect of CXCL10/IP-10 on oligodendrocyte progenitor cells appears to occur in patients with multiple sclerosis. Elevated CXCL1 expression occurs in areas of demyelination [61][63]. Research on oligodendrocyte precursors has shown that CXCL1 induces proliferation of these cells [4][56]. At the same time, this chemokine inhibits oligodendrocyte precursor cell migration [7][56]. As a consequence, oligodendrocyte precursor cells accumulate in areas of demyelination. Then, these cells participate in remyelination [56][57][58]. Finally, CXCL1 can inhibit multiple sclerosis by causing neutrophil infiltration into the brain. These cells are able to inhibit the activity of autoreactive T cells [75], which inhibits the progression of multiple sclerosis.
CXCL1 also participates in the progression of multiple sclerosis [61]. CXCR2 activation inhibits oligodendrocyte precursor cell differentiation, and so ligands of this receptor inhibit remyelination in this way [61]. CXCL1 also causes neutrophil infiltration into the central nervous system [69][71][72][78]. Neutrophils cause neurodegeneration, as they produce ROS that acts destructively on the neural tissue [79][80]. This effect is enhanced by CXCR2 activation [80]. These cells also produce proinflammatory cytokines and thus increase neuroinflammation in patients with multiple sclerosis. The cells described also cause a breakdown of the BBB [79]. Due to the importance of neutrophils in the course of multiple sclerosis, an elevated neutrophil-lymphocyte ratio (NLR) is associated with faster progression and a greater disability in patients with this disease [81][82], with neutrophils appearing in the cerebrospinal fluid of multiple sclerosis patients in the early stages of the disease [68]. Subsequently, the level of these cells decreases with treatment and length of the disease, which shows that neutrophils are an important pathogenic factor only in the initial stages of multiple sclerosis [68][69].
10. Neuromyelitis Optica
Neuromyelitis optica is a neurodegenerative and autoimmune disease. Auto-antibodies to aquaporin 4 (AQP4) are responsible in over 80% of cases of this disease [83]. The incidence of this disease is estimated to be between 0.5 and 10 cases per 100,000 population, depending on the country. These auto-antibodies directly cause an increase in CXCL1 secretion from astrocytes [84]. Therefore, CXCL1 levels are elevated in neuromyelitis optica in cerebrospinal fluid [67] and in serum [84]. It was found that CXCL1 levels are not correlated with patient clinical severity [67], which shows that CXCL1 can only be a marker for patients with neuromyelitis optica, but does not affect the development and severity of the disease. Also, studies in animal models have shown that CXCR2, the receptor for the chemokine in question, is irrelevant in the development of neuromyelitis optica [85]. However, studies in an animal model of neuromyelitis optica have shown that neutrophil proteases are an important factor in the pathophysiology of this disease [86]. CXCL1 is a chemokine that induces neutrophil recruitment. Therefore, it may play an indirect role in the development of neuromyelitis optica by recruiting these cells into the neural tissue.
11. Neuropathic Pain and Sickness Behaviors
Chemokines, such as CXCR2 ligands, are important in the development of neuropathic pain. However, due to bioethical constraints, all knowledge about the importance of CXC chemokines in the development of neuropathic pain is based on experiments on laboratory animals. Due to the fact that CXCR2 ligand systems differ significantly between humans and mice, the presented mechanism of neuropathic pain gives only a hint of the role that CXCL1 may play in this disease in humans.
Following nerve injury in mice, KC expression increases in spinal astrocytes [87][88][89]. This process is dependent on TNF-α which also increases the production of IL-1β. This pro-inflammatory cytokine increases cyclooxygenase-2 (COX-2) activity and the production of prostanoids which are also responsible for the sensation of pain [90]. KC also induces inflammatory responses, in particular the production of TNF-α, IL-1β, IL-6 and CCL2 [89][91]. Nerve injury is also associated with an increase in CXCR2 expression in dorsal horn neurons [87]. Similarly, following traumatic brain injury, increased CXCR2 expression has been observed in spinal cord neurons [92].
CXCR2 activation on neurons and spinal microglia has been found to lead to neuropathic pain [87][88][93][94]. Specifically, KC results in the release of sympathetic amines [90], increased signaling via the transient receptor potential vanilloid type 1 (TRPV1) channel on sensory neurons [95], upregulation of Na+ currents [96] and upregulation of K+ currents in sensory neurons [97]. Another mechanism by which CXCR2 ligands cause neuropathic pain is through increased release of calcitonin gene-related peptide (CGRP), as confirmed by studies on chemokine CINC-1 in rats [98]. CGRP is thus responsible for pain generation. Also, CXCR2-dependent KC increases N-methyl-D-aspartate (NMDA)-induced currents in lamina II neurons in a mouse model [88].
It has been found that KC can also cause neuropathic pain by recruiting neutrophils [89][99][100]. These cells secrete cathepsin E which is involved in pain [100]. However, activation of CXCR2 on neutrophils results in the release of endogenous opioids from these cells that reduce pain [101]. KC is also important in the formation of post-surgical pain, as infiltration of neutrophils near surgical wounds, cells that cause post-surgical pain, is dependent on the KC→CXCR2 axis in mice [102].
Also, studies with laboratory animals show that CXCR2 ligands reduce spontaneous activity [103]. This indicates that CXCL1 may be associated with poor mental status and sickness behaviors of patients with multiple sclerosis, as well as other neuroinflammatory diseases.
12. Prion Diseases
Prion diseases form a group of neurodegenerative diseases caused by scrapie-associated prion protein (PrPSc) [104]. This protein has a pathogenic misfolding that replicates by converting the structure of cellular prion protein (PrPC) into PrPSc. This is followed by aggregation of PrPSc in neural tissue which leads to spongiform degeneration. Examples of prion diseases include sporadic Creutzfeldt-Jakob disease (sCJD), fatal familial insomnia and kuru. An important element in the course of prion diseases is neuroinflammation [105] involving microglia and astroglia [106][107][108]. Studies in mice have shown that both cell types produce p40 subunit of IL-12 (IL-12p40) and CXCL10/IP-10 [106]. In contrast, astroglia produce cytokines such as IL-1β, IL-6, IL-12p70, CCL2, CCL3, CCL5 and KC.
As KC is a murine paralog for human CXCL1 [3][32], it may be suspected that humans with prion diseases have an increase in CXCL1 expression in the brain, although that needs to be confirmed by further research. Researchers also need more research on the significance of CXCL1 in the course of spongiform degeneration of the brain.
CXCL1 does not play an important role in prion diseases. In multiple sclerosis, an increase in CXCL1 expression in the brain has a destructive effect on neural tissue through infiltration of neural tissue by neutrophils [62][80]. Even when infiltration of neutrophils occurs in prion diseases, PrPSc reduces the activity of these cells [109] and therefore, these cells do not have a destructive effect on neural tissue and at the same time have no function in the course of prion diseases. Thus, the main property of CXCL1, which is the effect on neutrophils, does not play an important role in the course of prion diseases.
13. Tick-Borne Encephalitis (TBE) and Ticks
Tick-borne encephalitis (TBE) is a disease caused by the tick-borne encephalitis virus (TBEV) [110]. Its genetic material is a positive-sense single-stranded RNA of approximately 11kb in length. This virus belongs to the genus Flavivirus. It infects animals in central and eastern Europe and northern Asia. In humans, TBEV infection usually gives mild symptoms; however, some 2% of cases of TBEV infection are fatal. TBEV is a neurotropic pathogen that attacks neural tissue, including the brain. The vector for this virus is the Ixodes sp. tick, which means the virus enters the body of the host during feeding by these parasites.
At the same time, the feeding of the tick involves a strong immune response [111]. The tick’s bite initially induces an increased expression of pro-inflammatory cytokines that are chemoattractants for lymphocytes, macrophages and neutrophils (Figure 4). However, the expression of proinflammatory cytokines is downregulated [111] and chemokines are inactivated by tick salivary compounds [112]. Among these compounds are evasins [113][114][115][116], glycoproteins commonly found in the saliva of various tick species [114]. In particular, evasin-3 specifically binds to CXC chemokines, including CXCL1 [113]. CXCL1 in such a complex cannot activate its CXCR2 receptor but can still be bound to glycosaminoglycans [116]. This results in the inactivation of CXCL1 and reduced infiltration of the tick feeding site by neutrophils [113][115].
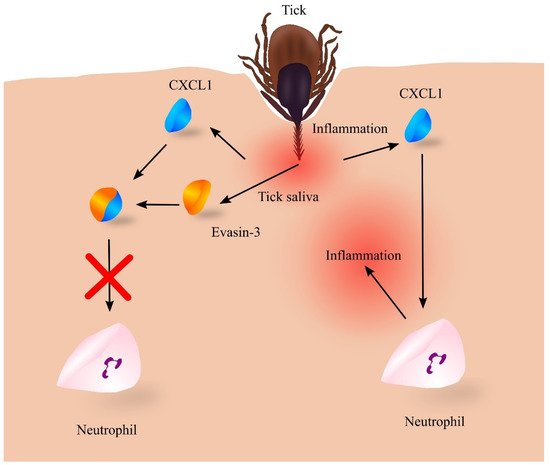
Figure 4. Role of evasin-3 in inhibiting the inflammatory response to tick feeding. Tick feeding leads to inflammatory responses, which induce an increase in the expression of chemokines including CXC motif chemokine ligand 1 (CXCL1), which recruits neutrophils to the vicinity of the feeding tick. This leads to even greater inflammatory reactions, itching of the skin, and removal of the tick by the host. However, the tick introduces its saliva into the site of the puncture while feeding, which contains a number of proteins, including evasin-3, which binds to CXCL1, resulting in the loss of its biological properties. No neutrophil recruitment then occurs and the tick can feed unnoticed for some time. Source: own elaboration.
Other evasins inactivate other groups of chemokines. For example, Evasin-1 inactivates the proinflammatory chemokines CCL3 and CCL4 [113]. Evasins and other tick salivary compounds are able to inhibit the immune response at the tick feeding site [112][115]. For this reason, the tick can feed unnoticed by its host for a prolonged time.
TBEV is a neurotropic pathogen causing a severe neuropathological disorder [110]. One component of this disease is neuroinflammation [117], associated with elevated levels of CXCL1 and CXCL8/IL-8 in cerebrospinal fluid [118]. The levels of these chemokines are correlated with neutrophil infiltration of neural tissue [118]. There is also an increase in IL-17 levels [118], a cytokine that increases CXCL1 expression [119][120]. This indicates that TBEV infection of neural tissue cells induces an increase in IL-17 production, which then causes an increase in CXCL1 recruiting neutrophils to the sites of the TBEV infection to control the disease.
14. Traumatic Spinal Cord Injury
Traumatic spinal cord injury involves damage to the spinal cord as a result of a violent accident or fall. It is estimated that there are approximately 17,000 cases of traumatic spinal cord injury per year in the United States alone [121]. Approximately one week after a traumatic spinal cord injury, patients experience an increase in blood CXCL1 levels [122]. This is the result of an increase in the expression of IL-1β in the liver, a pro-inflammatory cytokine which increases the expression of CXCL1 in this organ [123], triggering the mobilization of neutrophils from the bone marrow [124]. A similar mechanism occurs at the site of the spinal cord injury, where an increase in IL-1β production elevates CXCL1 expression [125][126]. This increased expression of CXCL1 in the spinal cord is not TNF-α dependent [125]. These processes are followed by infiltration of the spinal cord by neutrophils [123]. These cells secrete elastase, which causes additional damage to the spinal cord [127]. This leads to various secondary dysfunctions in patients with traumatic spinal cord injury.
15. West Nile Fever
West Nile fever is caused by West Nile Virus (WNV) from the genus Flavivirus [128]. Its genetic material is positive-sense single-stranded RNA with a length of about 11 kb. WNV is not a dangerous pathogen for humans with a healthy immune system where it causes only mild symptoms [128].
WNV acts as a neurotropic pathogen that affects the nervous system and also other tissues and organs such as the skin, kidney and gastrointestinal tract. WNV is a zoonotic pathogen that is transmitted to humans by mosquitoes, mainly by Culex sp. For this reason it is found in warm countries in Africa, Asia, Australia and Europe. When bitten by the mosquito, the virus is introduced into the skin [129]. It infects Langerhans cells and keratinocytes, where it replicates [128], causing inflammatory responses such as increased expression of pro-inflammatory cytokines and chemokines recruiting neutrophils: CXCL1, CXCL2 and CXCL8/IL-8 [129]. This reaction of the immune system results in the eradication of WNV infection.
Anti-inflammatory substances in the mosquito saliva reduce inflammatory responses [129], which allows WNV replication in the skin in the first stage of infection. If the infection is not suppressed by the immune system, WNV then enters the lymph nodes where it further replicates [128]. WNV causes increased expression of chemokines that attract neutrophils, including CXCL1 [130]. This is followed by the recruitment of neutrophils to the sites of WNV infection where these cells are also infected [130]. The virus replicates in neutrophils which then enter the blood and constitute a reservoir and a type of vector that causes dissemination of WNV throughout the body. However, at the late stages of infection, non-infected neutrophils are an important part of the fight against WNV infection [130].
WNV is a neurotropic pathogen that can get into the brain and spinal cord, although this form of infection is rare in humans [128]. There are many theories, some of which are mutually exclusive, about the mechanism by which WNV enters the neural tissue [128]. One of them suggests that WNV-infected neutrophils migrate into the neural tissue in a kind of ‘Trojan horse’ mechanism [131]. In the neural tissue, WNV infects neurons and astrocytes but not microglia cells [132][133], resulting in the death of infected cells. Then microglial cells phagocytose infected cells and cell debris [134] which causes the activation of the microglia and thus secretion of many pro-inflammatory cytokines and chemokines, including CXCL1 [134]. This leads to the infiltration of infected neural tissue by immune cells including neutrophils [135], resulting in the eradication of WNV infection.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23084205
References
- Bhardwaj, D.; Náger, M.; Camats, J.; David, M.; Benguria, A.; Dopazo, A.; Cantí, C.; Herreros, J. Chemokines induce axon outgrowth downstream of Hepatocyte Growth Factor and TCF/β-catenin signaling. Front. Cell. Neurosci. 2013, 7, 52.
- Deftu, A.T.; Ciorescu, R.; Gheorghe, R.O.; Mihăilescu, D.; Ristoiu, V. CXCL1 and CXCL2 Inhibit the Axon Outgrowth in a Time- and Cell-Type-Dependent Manner in Adult Rat Dorsal Root Ganglia Neurons. Neurochem. Res. 2019, 44, 2215–2229.
- Nomiyama, H.; Mera, A.; Ohneda, O.; Miura, R.; Suda, T.; Yoshie, O. Organization of the chemokine genes in the human and mouse major clusters of CC and CXC chemokines: Diversification between the two species. Genes Immun. 2001, 2, 110–113.
- Robinson, S.; Tani, M.; Strieter, R.M.; Ransohoff, R.M.; Miller, R.H. The chemokine growth-regulated oncogene-alpha promotes spinal cord oligodendrocyte precursor proliferation. J. Neurosci. 1998, 18, 10457–10463.
- Vora, P.; Pillai, P.; Mustapha, J.; Kowal, C.; Shaffer, S.; Bose, R.; Namaka, M.; Frost, E.E. CXCL1 regulation of oligodendrocyte progenitor cell migration is independent of calcium signaling. Exp. Neurol. 2012, 236, 259–267.
- Filipovic, R.; Zecevic, N. The effect of CXCL1 on human fetal oligodendrocyte progenitor cells. Glia 2008, 56, 1–15.
- Tsai, H.H.; Frost, E.; To, V.; Robinson, S.; Ffrench-Constant, C.; Geertman, R.; Ransohoff, R.M.; Miller, R.H. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell 2002, 110, 373–383.
- Huang, F.; Lan, Y.; Qin, L.; Dong, H.; Shi, H.; Wu, H.; Zou, Q.; Hu, Z.; Wu, X. Astragaloside IV Promotes Adult Neurogenesis in Hippocampal Dentate Gyrus of Mouse through CXCL1/CXCR2 Signaling. Molecules 2018, 23, 2178.
- Zonis, S.; Breunig, J.J.; Mamelak, A.; Wawrowsky, K.; Bresee, C.; Ginzburg, N.; Chesnokova, V. Inflammation-induced Gro1 triggers senescence in neuronal progenitors: Effects of estradiol. J. Neuroinflamm. 2018, 15, 260.
- Shang, Y.; Tian, L.; Chen, T.; Liu, X.; Zhang, J.; Liu, D.; Wei, J.; Fang, W.; Chen, Y.; Shang, D. CXCL1 promotes the proliferation of neural stem cells by stimulating the generation of reactive oxygen species in APP/PS1 mice. Biochem. Biophys. Res. Commun. 2019, 515, 201–206.
- Gordon, R.J.; McGregor, A.L.; Connor, B. Chemokines direct neural progenitor cell migration following striatal cell loss. Mol. Cell. Neurosci. 2009, 41, 219–232.
- Saika, F.; Matsuzaki, S.; Kobayashi, D.; Kiguchi, N.; Kishioka, S. Chemokine CXCL1 is responsible for cocaine-induced reward in mice. Neuropsychopharmacol. Rep. 2018, 38, 145–148.
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70.
- Craig-Schapiro, R.; Kuhn, M.; Xiong, C.; Pickering, E.H.; Liu, J.; Misko, T.P.; Perrin, R.J.; Bales, K.R.; Soares, H.; Fagan, A.M.; et al. Multiplexed immunoassay panel identifies novel CSF biomarkers for Alzheimer’s disease diagnosis and prognosis. PLoS ONE 2011, 6, e18850.
- Xia, M.; Hyman, B.T. GROalpha/KC, a chemokine receptor CXCR2 ligand, can be a potent trigger for neuronal ERK1/2 and PI-3 kinase pathways and for tau hyperphosphorylation-a role in Alzheimer’s disease? J. Neuroimmunol. 2002, 122, 55–64.
- Zhang, X.F.; Zhao, Y.F.; Zhu, S.W.; Huang, W.J.; Luo, Y.; Chen, Q.Y.; Ge, L.J.; Li, R.S.; Wang, J.F.; Sun, M.; et al. CXCL1 Triggers Caspase-3 Dependent Tau Cleavage in Long-Term Neuronal Cultures and in the Hippocampus of Aged Mice: Implications in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 89–104.
- Bakshi, P.; Margenthaler, E.; Laporte, V.; Crawford, F.; Mullan, M. Novel role of CXCR2 in regulation of gamma-secretase activity. ACS Chem. Biol. 2008, 3, 777–789.
- Bakshi, P.; Jin, C.; Broutin, P.; Berhane, B.; Reed, J.; Mullan, M. Structural optimization of a CXCR2-directed antagonist that indirectly inhibits gamma-secretase and reduces Abeta. Bioorg. Med. Chem. 2009, 17, 8102–8112.
- Bakshi, P.; Margenthaler, E.; Reed, J.; Crawford, F.; Mullan, M. Depletion of CXCR2 inhibits γ-secretase activity and amyloid-β production in a murine model of Alzheimer’s disease. Cytokine 2011, 53, 163–169.
- Zhang, K.; Tian, L.; Liu, L.; Feng, Y.; Dong, Y.B.; Li, B.; Shang, D.S.; Fang, W.G.; Cao, Y.P.; Chen, Y.H. CXCL1 contributes to β-amyloid-induced transendothelial migration of monocytes in Alzheimer’s disease. PLoS ONE 2013, 8, e72744.
- Simard, A.R.; Rivest, S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004, 18, 998–1000.
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701.
- Di Sapia, R.; Zimmer, T.S.; Kebede, V.; Balosso, S.; Ravizza, T.; Sorrentino, D.; Castillo, M.A.M.; Porcu, L.; Cattani, F.; Ruocco, A.; et al. CXCL1-CXCR1/2 signaling is induced in human temporal lobe epilepsy and contributes to seizures in a murine model of acquired epilepsy. Neurobiol. Dis. 2021, 158, 105468.
- Liu, X.X.; Yang, L.; Shao, L.X.; He, Y.; Wu, G.; Bao, Y.H.; Lu, N.N.; Gong, D.M.; Lu, Y.P.; Cui, T.T.; et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 2020, 217, e20180992.
- Kothur, K.; Bandodkar, S.; Wienholt, L.; Chu, S.; Pope, A.; Gill, D.; Dale, R.C. Etiology is the key determinant of neuroinflammation in epilepsy: Elevation of cerebrospinal fluid cytokines and chemokines in febrile infection-related epilepsy syndrome and febrile status epilepticus. Epilepsia 2019, 60, 1678–1688.
- Everett, R.D. HSV-1 biology and life cycle. Methods Mol. Biol. 2014, 1144, 1–17.
- McQuillan, G.; Kruszon-Moran, D.; Flagg, E.W.; Paulose-Ram, R. Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14–49: United States, 2015–2016. In NCHS Data Brief No. 304; National Center for Health Statistics: Hyattsville, MD, USA, 2018; pp. 1–8.
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820.
- Vilela, M.C.; Mansur, D.S.; Lacerda-Queiroz, N.; Rodrigues, D.H.; Arantes, R.M.; Kroon, E.G.; Campos, M.A.; Teixeira, M.M.; Teixeira, A.L. Traffic of leukocytes in the central nervous system is associated with chemokine up-regulation in a severe model of herpes simplex encephalitis: An intravital microscopy study. Neurosci. Lett. 2008, 445, 18–22.
- Michael, B.D.; Bricio-Moreno, L.; Sorensen, E.W.; Miyabe, Y.; Lian, J.; Solomon, T.; Kurt-Jones, E.A.; Luster, A.D. Astrocyte- and Neuron-Derived CXCL1 Drives Neutrophil Transmigration and Blood-Brain Barrier Permeability in Viral Encephalitis. Cell. Rep. 2020, 32, 108150.
- Nomiyama, H.; Osada, N.; Yoshie, O. Systematic classification of vertebrate chemokines based on conserved synteny and evolutionary history. Genes Cells 2013, 18, 1–16.
- Bozic, C.R.; Kolakowski, L.F., Jr.; Gerard, N.P.; Garcia-Rodriguez, C.; von Uexkull-Guldenband, C.; Conklyn, M.J.; Breslow, R.; Showell, H.J.; Gerard, C. Expression and biologic characterization of the murine chemokine KC. J. Immunol. 1995, 154, 6048–6057.
- West, D.M.; Del Rosso, C.R.; Yin, X.T.; Stuart, P.M. CXCL1 but not IL-6 is required for recurrent herpetic stromal keratitis. J. Immunol. 2014, 192, 1762–1767.
- Suryawanshi, A.; Veiga-Parga, T.; Reddy, P.B.; Rajasagi, N.K.; Rouse, B.T. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J. Immunol. 2012, 188, 3434–3446.
- Yan, X.T.; Tumpey, T.M.; Kunkel, S.L.; Oakes, J.E.; Lausch, R.N. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1854–1862.
- Bryant-Hudson, K.M.; Carr, D.J. CXCL1-deficient mice are highly sensitive to pseudomonas aeruginosa but not herpes simplex virus type 1 corneal infection. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6785–6792.
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142.
- Park, H.J.; Yun, D.H.; Kim, S.K.; Chung, J.H.; Lee, J.S.; Park, H.K.; Chon, J.; Kim, D.H.; Yoo, S.D.; Kim, H.S. Association of CXCL1 promoter polymorphism with ischaemic stroke in Korean population. Int. J. Immunogenet. 2013, 40, 306–310.
- Losy, J.; Zaremba, J.; Skrobański, P. CXCL1 (GRO-alpha) chemokine in acute ischaemic stroke patients. Folia Neuropathol. 2005, 43, 97–102.
- Gelderblom, M.; Weymar, A.; Bernreuther, C.; Velden, J.; Arunachalam, P.; Steinbach, K.; Orthey, E.; Arumugam, T.V.; Leypoldt, F.; Simova, O.; et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood 2012, 120, 3793–3802.
- Amin, M.; Vakilian, A.; Mahmoodi, M.H.; Hassanshahi, G.; Falahati-Pour, S.K.; Dolatabadi, M.R.; Nadimi, A.E. Circulatory Levels of C-X-C Motif Chemokine Ligands 1, 9, and 10 Are Elevated in Patients with Ischemic Stroke. Eurasian J. Med. 2017, 49, 92–96.
- Zhu, W.; Nan, Y.; Wang, S.; Liu, W. Bioinformatics Analysis of Gene Expression Profiles of Sex Differences in Ischemic Stroke. Biomed. Res. Int. 2019, 2019, 2478453.
- Shi, Y.; Yi, Z.; Zhao, P.; Xu, Y.; Pan, P. MicroRNA-532-5p protects against cerebral ischemia-reperfusion injury by directly targeting CXCL1. Aging 2021, 13, 11528–11541.
- Leng, J.; Liu, W.; Li, L.; Wei, F.Y.; Tian, M.; Liu, H.M.; Guo, W. MicroRNA-429/Cxcl1 Axis Protective Against Oxygen Glucose Deprivation/Reoxygenation-Induced Injury in Brain Microvascular Endothelial Cells. Dose Response 2020, 18, 1559325820913785.
- Brait, V.H.; Rivera, J.; Broughton, B.R.; Lee, S.; Drummond, G.R.; Sobey, C.G. Chemokine-related gene expression in the brain following ischemic stroke: No role for CXCR2 in outcome. Brain Res. 2011, 1372, 169–179.
- Ikegame, Y.; Yamashita, K.; Hayashi, S.; Yoshimura, S.; Nakashima, S.; Iwama, T. Neutrophil elastase inhibitor prevents ischemic brain damage via reduction of vasogenic edema. Hypertens. Res. 2010, 33, 703–707.
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312.
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006.
- Song, A.Q.; Gao, B.; Fan, J.J.; Zhu, Y.J.; Zhou, J.; Wang, Y.L.; Xu, L.Z.; Wu, W.N. NLRP1 inflammasome contributes to chronic stress-induced depressive-like behaviors in mice. J. Neuroinflamm. 2020, 17, 178.
- Pandey, G.N.; Rizavi, H.S.; Bhaumik, R.; Zhang, H. Chemokines gene expression in the prefrontal cortex of depressed suicide victims and normal control subjects. Brain Behav. Immun. 2021, 94, 266–273.
- Bot, M.; Chan, M.K.; Jansen, R.; Lamers, F.; Vogelzangs, N.; Steiner, J.; Leweke, F.M.; Rothermundt, M.; Cooper, J.; Bahn, S.; et al. Serum proteomic profiling of major depressive disorder. Transl. Psychiatry 2015, 5, e599.
- Fanelli, G.; Benedetti, F.; Wang, S.M.; Lee, S.J.; Jun, T.Y.; Masand, P.S.; Patkar, A.A.; Han, C.; Serretti, A.; Pae, C.U.; et al. Reduced CXCL1/GRO chemokine plasma levels are a possible biomarker of elderly depression. J. Affect. Disord. 2019, 249, 410–417.
- Walss-Bass, C.; Suchting, R.; Olvera, R.L.; Williamson, D.E. Inflammatory markers as predictors of depression and anxiety in adolescents: Statistical model building with component-wise gradient boosting. J. Affect. Disord. 2018, 234, 276–281.
- Lee, K.S.; Chung, J.H.; Lee, K.H.; Shin, M.J.; Oh, B.H.; Lee, S.H.; Hong, C.H. Simultaneous measurement of 23 plasma cytokines in late-life depression. Neurol. Sci. 2009, 30, 435–438.
- Leighton, S.P.; Nerurkar, L.; Krishnadas, R.; Johnman, C.; Graham, G.J.; Cavanagh, J. Chemokines in depression in health and in inflammatory illness: A systematic review and meta-analysis. Mol. Psychiatry 2018, 23, 48–58.
- Omari, K.M.; Lutz, S.E.; Santambrogio, L.; Lira, S.A.; Raine, C.S. Neuroprotection and remyelination after autoimmune demyelination in mice that inducibly overexpress CXCL1. Am. J. Pathol. 2009, 174, 164–176.
- Omari, K.M.; John, G.R.; Sealfon, S.C.; Raine, C.S. CXC chemokine receptors on human oligodendrocytes: Implications for multiple sclerosis. Brain 2005, 128, 1003–1015.
- Karim, H.; Kim, S.H.; Lapato, A.S.; Yasui, N.; Katzenellenbogen, J.A.; Tiwari-Woodruff, S.K. Increase in chemokine CXCL1 by ERβ ligand treatment is a key mediator in promoting axon myelination. Proc. Natl. Acad. Sci. USA 2018, 115, 6291–6296.
- Howard, J.; Trevick, S.; Younger, D.S. Epidemiology of Multiple Sclerosis. Neurol. Clin. 2016, 34, 919–939.
- Garg, N.; Smith, T.W. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015, 5, e00362.
- Kerstetter, A.E.; Padovani-Claudio, D.A.; Bai, L.; Miller, R.H. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp. Neurol. 2009, 220, 44–56.
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35.
- Dulamea, A.O. Role of Oligodendrocyte Dysfunction in Demyelination, Remyelination and Neurodegeneration in Multiple Sclerosis. Adv. Exp. Med. Biol. 2017, 958, 91–127.
- Burman, J.; Svensson, E.; Fransson, M.; Loskog, A.S.; Zetterberg, H.; Raininko, R.; Svenningsson, A.; Fagius, J.; Mangsbo, S.M. The cerebrospinal fluid cytokine signature of multiple sclerosis: A homogenous response that does not conform to the Th1/Th2/Th17 convention. J. Neuroimmunol. 2014, 277, 153–159.
- Khaibullin, T.; Ivanova, V.; Martynova, E.; Cherepnev, G.; Khabirov, F.; Granatov, E.; Rizvanov, A.; Khaiboullina, S. Elevated Levels of Proinflammatory Cytokines in Cerebrospinal Fluid of Multiple Sclerosis Patients. Front. Immunol. 2017, 8, 531.
- Lepennetier, G.; Hracsko, Z.; Unger, M.; Van Griensven, M.; Grummel, V.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Kowarik, M.C. Cytokine and immune cell profiling in the cerebrospinal fluid of patients with neuro-inflammatory diseases. J. Neuroinflamm. 2019, 16, 219.
- Liu, Z.; Chen, J.; Wang, Z.; Wang, Y.; Zheng, D.; Wang, H.; Peng, Y. The CSF Levels of Neutrophil-Related Chemokines in Patients with Neuromyelitis Optica. Ann. Clin. Transl. Neurol. 2020, 7, 1245–1251.
- Kostic, M.; Dzopalic, T.; Zivanovic, S.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I.; Vojinovic, S.; Marjanovic, G.; Savic, V.; Colic, M. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand. J. Immunol. 2014, 79, 181–186.
- Wojkowska, D.W.; Szpakowski, P.; Ksiazek-Winiarek, D.; Leszczynski, M.; Glabinski, A. Interactions between neutrophils, Th17 cells, and chemokines during the initiation of experimental model of multiple sclerosis. Mediat. Inflamm. 2014, 2014, 590409.
- Filipovic, R.; Jakovcevski, I.; Zecevic, N. GRO-alpha and CXCR2 in the human fetal brain and multiple sclerosis lesions. Dev. Neurosci. 2003, 25, 279–290.
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823.
- Simmons, S.B.; Liggitt, D.; Goverman, J.M. Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 2014, 193, 555–563.
- Kang, Z.; Wang, C.; Zepp, J.; Wu, L.; Sun, K.; Zhao, J.; Chandrasekharan, U.; DiCorleto, P.E.; Trapp, B.D.; Ransohoff, R.M.; et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat. Neurosci. 2013, 16, 1401–1408.
- Miller, N.M.; Wang, J.; Tan, Y.; Dittel, B.N. Anti-inflammatory mechanisms of IFN-γ studied in experimental autoimmune encephalomyelitis reveal neutrophils as a potential target in multiple sclerosis. Front. Neurosci. 2015, 9, 287.
- Zehntner, S.P.; Brickman, C.; Bourbonnière, L.; Remington, L.; Caruso, M.; Owens, T. Neutrophils that infiltrate the central nervous system regulate T cell responses. J. Immunol. 2005, 174, 5124–5131.
- Tirotta, E.; Kirby, L.A.; Hatch, M.N.; Lane, T.E. IFN-γ-induced apoptosis of human embryonic stem cell derived oligodendrocyte progenitor cells is restricted by CXCR2 signaling. Stem Cell Res. 2012, 9, 208–217.
- Tirotta, E.; Ransohoff, R.M.; Lane, T.E. CXCR2 signaling protects oligodendrocyte progenitor cells from IFN-γ/CXCL10-mediated apoptosis. Glia 2011, 59, 1518–1528.
- Grist, J.J.; Marro, B.S.; Skinner, D.D.; Syage, A.R.; Worne, C.; Doty, D.J.; Fujinami, R.S.; Lane, T.E. Induced CNS expression of CXCL1 augments neurologic disease in a murine model of multiple sclerosis via enhanced neutrophil recruitment. Eur. J. Immunol. 2018, 48, 1199–1210.
- De Bondt, M.; Hellings, N.; Opdenakker, G.; Struyf, S. Neutrophils: Underestimated Players in the Pathogenesis of Multiple Sclerosis (MS). Int. J. Mol. Sci. 2020, 21, 4558.
- Khaw, Y.M.; Cunningham, C.; Tierney, A.; Sivaguru, M.; Inoue, M. Neutrophil-selective deletion of Cxcr2 protects against CNS neurodegeneration in a mouse model of multiple sclerosis. J. Neuroinflamm. 2020, 17, 49.
- D’Amico, E.; Zanghì, A.; Romano, A.; Sciandra, M.; Palumbo, G.A.M.; Patti, F. The Neutrophil-to-Lymphocyte Ratio is Related to Disease Activity in Relapsing Remitting Multiple Sclerosis. Cells 2019, 8, 1114.
- Hemond, C.C.; Glanz, B.I.; Bakshi, R.; Chitnis, T.; Healy, B.C. The neutrophil-to-lymphocyte and monocyte-to-lymphocyte ratios are independently associated with neurological disability and brain atrophy in multiple sclerosis. BMC Neurol. 2019, 19, 23.
- Jarius, S.; Paul, F.; Weinshenker, B.G.; Levy, M.; Kim, H.J.; Wildemann, B. Neuromyelitis optica. Nat. Rev. Dis. Primers 2020, 6, 85.
- Howe, C.L.; Kaptzan, T.; Magaña, S.M.; Ayers-Ringler, J.R.; LaFrance-Corey, R.G.; Lucchinetti, C.F. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia 2014, 62, 692–708.
- Jones, M.V.; Levy, M. Effect of CXCR2 Inhibition on Behavioral Outcomes and Pathology in Rat Model of Neuromyelitis Optica. J. Immunol. Res. 2018, 2018, 9034695.
- Saadoun, S.; Waters, P.; MacDonald, C.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann. Neurol. 2012, 71, 323–333.
- Zhang, Z.J.; Cao, D.L.; Zhang, X.; Ji, R.R.; Gao, Y.J. Chemokine contribution to neuropathic pain: Respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain 2013, 154, 2185–2197.
- Cao, D.L.; Zhang, Z.J.; Xie, R.G.; Jiang, B.C.; Ji, R.R.; Gao, Y.J. Chemokine CXCL1 enhances inflammatory pain and increases NMDA receptor activity and COX-2 expression in spinal cord neurons via activation of CXCR2. Exp. Neurol. 2014, 261, 328–336.
- Manjavachi, M.N.; Costa, R.; Quintão, N.L.; Calixto, J.B. The role of keratinocyte-derived chemokine (KC) on hyperalgesia caused by peripheral nerve injury in mice. Neuropharmacology 2014, 79, 17–27.
- Cunha, T.M.; Verri, W.A., Jr.; Silva, J.S.; Poole, S.; Cunha, F.Q.; Ferreira, S.H. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 1755–1760.
- Zhou, W.; Zhou, Y.; Wang, M.; Qian, C.; Wang, C.; Tang, J.; Cai, Z.; Dai, W.; Zhu, X. Pharmacological inhibition of CXCR2 alleviates neuropathic pain by inactivating microglia in a rat L5 spinal nerve ligation model. Am. J. Transl. Res. 2020, 12, 3803–3812.
- Liang, D.Y.; Shi, X.; Liu, P.; Sun, Y.; Sahbaie, P.; Li, W.W.; Yeomans, D.C.; Clark, J.D. The Chemokine Receptor CXCR2 Supports Nociceptive Sensitization after Traumatic Brain Injury. Mol. Pain 2017, 13, 1744806917730212.
- Manjavachi, M.N.; Quintão, N.L.; Campos, M.M.; Deschamps, I.K.; Yunes, R.A.; Nunes, R.J.; Leal, P.C.; Calixto, J.B. The effects of the selective and non-peptide CXCR2 receptor antagonist SB225002 on acute and long-lasting models of nociception in mice. Eur. J. Pain 2010, 14, 23–31.
- Moraes, T.R.; Elisei, L.S.; Malta, I.H.; Galdino, G. Participation of CXCL1 in the glial cells during neuropathic pain. Eur. J. Pharmacol. 2020, 875, 173039.
- Dong, F.; Du, Y.R.; Xie, W.; Strong, J.A.; He, X.J.; Zhang, J.M. Increased function of the TRPV1 channel in small sensory neurons after local inflammation or in vitro exposure to the pro-inflammatory cytokine GRO/KC. Neurosci. Bull. 2012, 28, 155–164.
- Wang, J.G.; Strong, J.A.; Xie, W.; Yang, R.H.; Coyle, D.E.; Wick, D.M.; Dorsey, E.D.; Zhang, J.M. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Mol. Pain 2008, 4, 38.
- Yang, R.H.; Strong, J.A.; Zhang, J.M. NF-kappaB mediated enhancement of potassium currents by the chemokine CXCL1/growth related oncogene in small diameter rat sensory neurons. Mol. Pain 2009, 5, 26.
- Qin, X.; Wan, Y.; Wang, X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J. Neurosci. Res. 2005, 82, 51–62.
- Cao, L.; Malon, J.T. Anti-nociceptive Role of CXCL1 in a Murine Model of Peripheral Nerve Injury-induced Neuropathic Pain. Neuroscience 2018, 372, 225–236.
- Harada, Y.; Zhang, J.; Imari, K.; Yamasaki, R.; Ni, J.; Wu, Z.; Yamamoto, K.; Kira, J.I.; Nakanishi, H.; Hayashi, Y. Cathepsin E in neutrophils contributes to the generation of neuropathic pain in experimental autoimmune encephalomyelitis. Pain 2019, 160, 2050–2062.
- Rittner, H.L.; Labuz, D.; Schaefer, M.; Mousa, S.A.; Schulz, S.; Schäfer, M.; Stein, C.; Brack, A. Pain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cells. FASEB J. 2006, 20, 2627–2629.
- Carreira, E.U.; Carregaro, V.; Teixeira, M.M.; Moriconi, A.; Aramini, A.; Verri, W.A., Jr.; Ferreira, S.H.; Cunha, F.Q.; Cunha, T.M. Neutrophils recruited by CXCR1/2 signalling mediate post-incisional pain. Eur. J. Pain 2013, 17, 654–663.
- Campbell, S.J.; Meier, U.; Mardiguian, S.; Jiang, Y.; Littleton, E.T.; Bristow, A.; Relton, J.; Connor, T.J.; Anthony, D.C. Sickness behaviour is induced by a peripheral CXC-chemokine also expressed in multiple sclerosis and EAE. Brain Behav. Immun. 2010, 24, 738–746.
- Sigurdson, C.J.; Bartz, J.C.; Glatzel, M. Cellular and Molecular Mechanisms of Prion Disease. Annu. Rev. Pathol. 2019, 14, 497–516.
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65.
- Tribouillard-Tanvier, D.; Striebel, J.F.; Peterson, K.E.; Chesebro, B. Analysis of protein levels of 24 cytokines in scrapie agent-infected brain and glial cell cultures from mice differing in prion protein expression levels. J. Virol. 2009, 83, 11244–11253.
- Tribouillard-Tanvier, D.; Race, B.; Striebel, J.F.; Carroll, J.A.; Phillips, K.; Chesebro, B. Early cytokine elevation, PrPres deposition, and gliosis in mouse scrapie: No effect on disease by deletion of cytokine genes IL-12p40 and IL-12p35. J. Virol. 2012, 86, 10377–10383.
- Hennessy, E.; Griffin, É.W.; Cunningham, C. Astrocytes Are Primed by Chronic Neurodegeneration to Produce Exaggerated Chemokine and Cell Infiltration Responses to Acute Stimulation with the Cytokines IL-1β and TNF-α. J. Neurosci. 2015, 35, 8411–8422.
- Miragliotta, G.; Fumarulo, R.; Fumarola, D. Inhibition of neutrophil functions by scrapie prion protein: Description of some inhibitory properties. Acta Virol. 1990, 34, 517–522.
- Ruzek, D.; Avšič Županc, T.; Borde, J.; Chrdle, A.; Eyer, L.; Karganova, G.; Kholodilov, I.; Knap, N.; Kozlovskaya, L.; Matveev, A.; et al. Tick-borne encephalitis in Europe and Russia: Review of pathogenesis, clinical features, therapy, and vaccines. Antivir. Res. 2019, 164, 23–51.
- Glatz, M.; Means, T.; Haas, J.; Steere, A.C.; Müllegger, R.R. Characterization of the early local immune response to Ixodes ricinus tick bites in human skin. Exp. Dermatol. 2017, 26, 263–269.
- Kazimírová, M.; Štibrániová, I. Tick salivary compounds: Their role in modulation of host defences and pathogen transmission. Front. Cell. Infect. Microbiol. 2013, 3, 43.
- Déruaz, M.; Frauenschuh, A.; Alessandri, A.L.; Dias, J.M.; Coelho, F.M.; Russo, R.C.; Ferreira, B.R.; Graham, G.J.; Shaw, J.P.; Wells, T.N.; et al. Ticks produce highly selective chemokine binding proteins with antiinflammatory activity. J. Exp. Med. 2008, 205, 2019–2031.
- Hayward, J.; Sanchez, J.; Perry, A.; Huang, C.; Rodriguez Valle, M.; Canals, M.; Payne, R.J.; Stone, M.J. Ticks from diverse genera encode chemokine-inhibitory evasin proteins. J. Biol. Chem. 2017, 292, 15670–15680.
- Lee, A.W.; Deruaz, M.; Lynch, C.; Davies, G.; Singh, K.; Alenazi, Y.; Eaton, J.R.O.; Kawamura, A.; Shaw, J.; Proudfoot, A.E.I.; et al. A knottin scaffold directs the CXC-chemokine-binding specificity of tick evasins. J. Biol. Chem. 2019, 294, 11199–11212.
- Denisov, S.S.; Heinzmann, A.C.A.; Vajen, T.; Vries, M.H.M.; Megens, R.T.A.; Suylen, D.; Koenen, R.R.; Post, M.J.; Ippel, J.H.; Hackeng, T.M.; et al. Tick Saliva Protein Evasin-3 Allows for Visualization of Inflammation in Arteries through Interactions with CXC-Type Chemokines Deposited on Activated Endothelium. Bioconjug. Chem. 2020, 31, 948–955.
- Pokorna Formanova, P.; Palus, M.; Salat, J.; Hönig, V.; Stefanik, M.; Svoboda, P.; Ruzek, D. Changes in cytokine and chemokine profiles in mouse serum and brain, and in human neural cells, upon tick-borne encephalitis virus infection. J. Neuroinflamm. 2019, 16, 205.
- Grygorczuk, S.; Świerzbińska, R.; Kondrusik, M.; Dunaj, J.; Czupryna, P.; Moniuszko, A.; Siemieniako, A.; Pancewicz, S. The intrathecal expression and pathogenetic role of Th17 cytokines and CXCR2-binding chemokines in tick-borne encephalitis. J. Neuroinflamm. 2018, 15, 115.
- Herjan, T.; Yao, P.; Qian, W.; Li, X.; Liu, C.; Bulek, K.; Sun, D.; Yang, W.P.; Zhu, J.; He, A.; et al. HuR is required for IL-17-induced Act1-mediated CXCL1 and CXCL5 mRNA stabilization. J. Immunol. 2013, 191, 640–649.
- Herjan, T.; Hong, L.; Bubenik, J.; Bulek, K.; Qian, W.; Liu, C.; Li, X.; Chen, X.; Yang, H.; Ouyang, S.; et al. IL-17-receptor-associated adaptor Act1 directly stabilizes mRNAs to mediate IL-17 inflammatory signaling. Nat. Immunol. 2018, 19, 354–365.
- Rabinstein, A.A. Traumatic Spinal Cord Injury. Continuum 2018, 24, 551–566.
- Hassanshahi, G.; Amin, M.; Shunmugavel, A.; Vazirinejad, R.; Vakilian, A.; Sanji, M.; Shamsizadeh, A.; RafatPanah, H.; Poor, N.M.; Moosavi, S.R.; et al. Temporal expression profile of CXC chemokines in serum of patients with spinal cord injury. Neurochem. Int. 2013, 63, 363–367.
- Yates, A.G.; Jogia, T.; Gillespie, E.R.; Couch, Y.; Ruitenberg, M.J.; Anthony, D.C. Acute IL-1RA treatment suppresses the peripheral and central inflammatory response to spinal cord injury. J. Neuroinflamm. 2021, 18, 15.
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593.
- Ellman, D.G.; Lund, M.C.; Nissen, M.; Nielsen, P.S.; Sørensen, C.; Lester, E.B.; Thougaard, E.; Jørgensen, L.H.; Nedospasov, S.A.; Andersen, D.C.; et al. Conditional Ablation of Myeloid TNF Improves Functional Outcome and Decreases Lesion Size after Spinal Cord Injury in Mice. Cells 2020, 9, 2407.
- Shiraishi, Y.; Kimura, A.; Kimura, H.; Ohmori, T.; Takahashi, M.; Takeshita, K. Deletion of inflammasome adaptor protein ASC enhances functional recovery after spinal cord injury in mice. J. Orthop. Sci. 2021, 26, 487–493.
- Tonai, T.; Shiba, K.; Taketani, Y.; Ohmoto, Y.; Murata, K.; Muraguchi, M.; Ohsaki, H.; Takeda, E.; Nishisho, T. A neutrophil elastase inhibitor (ONO-5046) reduces neurologic damage after spinal cord injury in rats. J. Neurochem. 2001, 78, 1064–1072.
- Habarugira, G.; Suen, W.W.; Hobson-Peters, J.; Hall, R.A.; Bielefeldt-Ohmann, H. West Nile Virus: An Update on Pathobiology, Epidemiology, Diagnostics, Control and “One Health” Implications. Pathogens 2020, 9, 589.
- Garcia, M.; Alout, H.; Diop, F.; Damour, A.; Bengue, M.; Weill, M.; Missé, D.; Lévêque, N.; Bodet, C. Innate Immune Response of Primary Human Keratinocytes to West Nile Virus Infection and Its Modulation by Mosquito Saliva. Front. Cell. Infect. Microbiol. 2018, 8, 387.
- Bai, F.; Kong, K.F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J. Infect. Dis. 2010, 202, 1804–1812.
- Paul, A.M.; Acharya, D.; Duty, L.; Thompson, E.A.; Le, L.; Stokic, D.S.; Leis, A.A.; Bai, F. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil “Trojan horse” transport. Sci. Rep. 2017, 7, 4722.
- Cheeran, M.C.; Hu, S.; Sheng, W.S.; Rashid, A.; Peterson, P.K.; Lokensgard, J.R. Differential responses of human brain cells to West Nile virus infection. J. Neurovirol. 2005, 11, 512–524.
- Hunsperger, E.; Roehrig, J.T. Characterization of West Nile viral replication and maturation in peripheral neurons in culture. J. Neurovirol. 2005, 11, 11–22.
- Quick, E.D.; Leser, J.S.; Clarke, P.; Tyler, K.L. Activation of intrinsic immune responses and microglial phagocytosis in an ex vivo spinal cord slice culture model of West Nile virus infection. J. Virol. 2014, 88, 13005–13014.
- Bréhin, A.C.; Mouriès, J.; Frenkiel, M.P.; Dadaglio, G.; Desprès, P.; Lafon, M.; Couderc, T. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+B220−BST-2+ leukocyte population. J. Immunol. 2008, 180, 6760–6767.
This entry is offline, you can click here to edit this entry!