1. Gene Therapy for Acquired Cholestasis
Since no definitive treatment has yet been developed for some acquired hepatic cholestasis, such as PBC and PSC, there is a great need to identify novel therapeutic alternatives that can reduce fibrogenesis and potentially prevent the development of chronic liver injury, making genetic-based treatments an attractive strategy to achieve sustained long-term therapeutic effects.
To generate animal models of acquired cholestatic disorders, interventions including bile duct ligation (BDL) and the induction of cholestasis by drugs, such as estrogens and carbon tetrachloride (CCL
4), have been utilized [
92]. The development of cholestasis involves several processes including: cellular apoptosis, production of proinflammatory cytokines, and fibrogenesis that ultimately leads to biliary impairment [
93].
Gene therapy approaches for acquired cholestasis have been addressed to mitigate liver damage by reducing apoptosis and fibrosis and improving bile formation (
Figure 3).
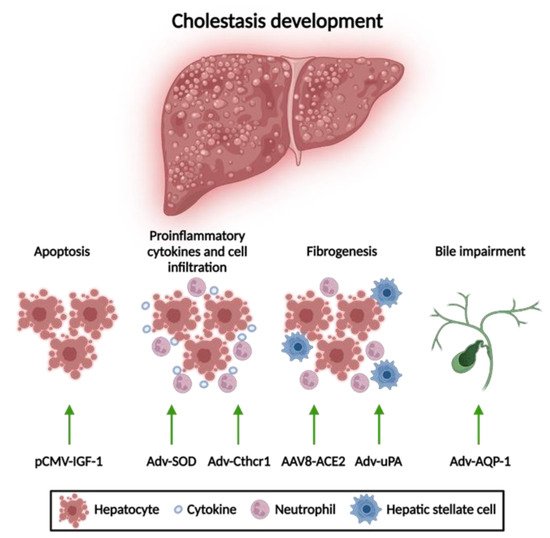
Figure 3. Gene therapy approaches for acquired cholestatic diseases. Different gene therapy strategies have resulted in an alleviation of liver disorders according to their anti-apoptotic, anti-inflammatory, and anti-fibrotic properties, respectively. Adv, adenoviral vector; AAV8, adeno-associated vector with serotype 8; ACE2, angiotensin-converting enzyme; AQP-1, aquaporin; Cthrc-1, collagen triple helix repeat containing-1; HNF4a, hepatocyte nuclear factor 4 alpha; IGF, insulin-like growth factor; SOD, superoxide dismutase; uPA, urokinase-plasminogen activator. This figure was created using BioRender.com.
1.1. Apoptosis Attenuation
One of the main targets for gene therapy of acquired liver disorders is the reduction of hepatocyte apoptosis. Hydrodynamic-based gene delivery to the liver of an insulin-like growth factor 1 (IGF-1)-expressing plasmid has demonstrated attenuation of hepatocellular apoptosis and liver injury in rats with BDL. IGF-1 promotes amelioration of cholestatic disease through activation of the phosphatidylinositol-3-kinase pathway, the inhibition of glycogen synthase kinase-3 beta, and the blockade of caspase-9 cleavage. Additionally, inactivation of hepatic stellate cells has been observed, which may explain the notable improvement in the degree of liver fibrosis [
94].
1.2. Reduction of Mitochondrial Oxidative Stress
Reducing oxidative stress has been shown to be a therapeutic target for acquired liver cholestasis. For example, Adv-mediated mitochondrial superoxide dismutase (SOD) gene delivery leads to a reduction in liver injury by avoiding the formation of oxygen free radicals derived from the accumulation of hydrophobic BAs and preventing the release of proinflammatory cytokines, such as TNFα and TGF-β, in mice with BDL [
95]. Similarly, administration of Adv vectors expressing an inhibitor gene of proinflammatory cytokine signaling like collagen triple helix repeat containing-1 (Cthrc-1) has shown a reduction of liver fibrosis in mice subjected to BDL and drug-mediated cholestasis through the inhibition of TGF-β signaling caused by the accelerating degradation of phospho-Smad3 [
96].
1.3. Anti-Fibrotic Therapies
Anti-fibrotic therapies for cholestatic disorders via reducing pro-inflammatory factors tend to promote collagen degradation and thus reduce the degree of liver fibrosis. Adv vectors expressing the urokinase-plasminogen activator (uPA) gene resulted in a slight reduction of liver fibrosis, leading to a partial improvement of liver histology in rats with BDL associated with the activation of metalloproteinases that trigger collagen degradation [
97,
98]. Additionally, AAV vectors that allow hepatic expression of angiotensin-converting enzyme (ACE2) provided a sustained anti-fibrotic effect in different animal models of BDL and drug-induced cholestasis [
99]. A different strategy to fight fibrosis is based on the gene delivery of human hepatocyte nuclear factor 4 alpha (HNF4A) via AAV vectors or mRNA containing LNP. This type of gene therapy was able to decrease the expression of genes involved in profibrogenic activity and revert fibrosis in several mouse models with induced or genetic cholestasis [
100].
1.4. Amelioration of Bile Flow
Finally, Adv-mediated hepatic delivery of aquaporin-1 (AQP1) has shown an improvement in the bile flow of estrogen-induced cholestatic rats [
101]. In fact, this approach resulted in a marked reduction of serum ALP, as well as serum and biliary concentrations of bile salts. Moreover, AQP1 gene transfer increased biliary output as mediated by a significant increase in BSEP transport activity [
102].
Thus, gene therapy approaches may offer a new avenue for the development of novel treatments for acquired cholestatic disorders.
2. Gene Therapy for Inherited Cholestasis
Gene therapy for the treatment of inherited hepatic diseases has garnered a great deal of attention after demonstrating that AAV vectors expressing human coagulation factors IX and VIII in the livers of patients with hemophilia B and A, respectively, resulted in a sustained therapeutic effect for more than three years [
103]. In fact, a large number of gene therapy products have demonstrated promising therapeutic effects in clinically relevant animal models, leading to clinical trials for inherited liver disorders, such as phenylketonuria, familial hypercholesterolemia, ornithine transcarbamylase deficiency, acute intermittent porphyria, methylmalonic acidemia, and Wilson’s disease, among others [
88].
2.1. Gene Therapy of Genetic Disorders with Associated Cholestasis
Preclinical studies have shown promising results in animal models of Cerebrotendinous xanthomatosis (CTX) and Crigler-Najjar syndrome type 1. In the first case, the administration of an AAV8 vector expressing CYP27A was able to restore BA metabolism and normalize the concentration of most BAs in plasma in a mouse model of CTX [
104]. Interestingly, this therapeutic effect was achieved with only 20% of transduced hepatocytes, which could greatly facilitate the clinical translation of this approach. Secondly, treatment of Crigler–Najjar syndrome type 1 with an AAV8 vector expressing UDP-glucuronosyltransferase family 1-member A1 (UGT1A) showed normalization of total serum bilirubin levels in two animal models of the disease, Gunn rats and
Ugt1a1-/- mice [
105]. In this last model, a therapeutic effect was also demonstrated in newborn mice, although high doses of vector were required to maintain the effect [
106]. These preclinical results led to a phase I/II clinical trial sponsored by Genethon (Évry, France), which is currently ongoing (NCT03466463).
The results observed in preclinical studies of Crigler–Najjar syndrome showed that one of the main limitations for gene therapy of genetic cholestatic diseases could be related to the loss of viral genomes associated with hepatocyte proliferation occurring in young patients [
107].
2.2. Gene Therapy for PFIC Diseases
Gene therapy approaches for PFIC can be based on gene supplementation or gene editing strategies to modify and repair the affected genes. The implementation of gene therapy for the different types of PFIC has some limitations. Firstly, in some types of PFIC in order to achieve stable and long-term therapeutic efficacy, it could be necessary to transduce most of the hepatocytes, which may require the use of high doses of the viral vector with the concomitant safety concerns [
107,
108]. Secondly, some types of PFIC have extrahepatic clinical manifestations hampering the liver-targeted treatment [
109]. Finally, PFIC diseases requiring therapy are generally diagnosed in pediatric patients, and gene therapy based on non-integrative vectors, such as AAV, may be inefficient due to the loss of viral genomes associated with hepatocyte proliferation in a growing liver [
107]. The decision to undergo gene therapy for PFIC, as well as the outcome of the therapy, will likely be influenced by the type of mutations present in the affected gene. For example, patients with missense mutations leading to decreased protein activity will probably respond better than those with a complete deficiency.
Although the loss of viral genomes could be a problem for most inherited cholestasis, ABCB4 deficiency, which causes PFIC3, has certain advantages over other PFIC types for liver gene therapy. For example, previous results using hepatocyte transplantation in a mouse model of PFIC3 showed that engraftment of 12% of healthy hepatocytes was enough to achieve therapeutic efficacy [
110]. This evidence led to four preclinical studies examining the feasibility of gene therapy for PFIC3 in three different
Abcb4-/- mouse models with a range of phenotypes depending on the mouse strain [
111].
Gene Therapy for PFIC3 Based on ABCB4 Supplementation
The first study tested gene therapy in C57BL/6
Abcb4-/- mice that were challenged with a BA-enriched diet to increase liver toxicity due to their mild phenotype. Treatment with an AAV8 vector expressing ABCB4 demonstrated long-term efficacy by preventing the increase of serum transaminases and the loss of biliary PC levels after BA challenge [
112]. The therapeutic effect was dose dependent, and it was observed that restoration of biliary PC levels above 12–13% (over 4000 µM) of wild-type levels was enough to have a curative effect. This indicates that PFIC3 could be treated even if only a small fraction of hepatocytes were transduced, in this way resembling gene therapy of other diseases like hemophilia B, in which therapeutic effects can be obtained with a small percentage of transduced hepatocytes.
Recently, a preclinical study based on LNP-encapsulated mRNA therapy was able to transiently reverse the disease phenotype in BALB/c
Abcb4-/- mice [
114]. BALB/c
Abcb4-/- show similar levels of serum biomarkers as the FVB
Abcb4-/- mice, but with a faster progression of liver fibrosis, leading to early development of primary liver cancers as well as an earlier onset of other complications, such as portal hypertension [
111]. Five repeat
ABCB4 mRNA-LNP injections were able to restore ABCB4 expression and biliary PC levels (~42% of wild-type levels), as well as improve serum biomarker levels, liver fibrosis, and hepatomegaly [
114,
115]. However, these previously described non-integrative vector-based gene therapy strategies may have important limitations, such as loss of transgene expression, either because of loss of viral genomes due to hepatocyte division or because the short half-life of mRNA requires periodic administration to maintain the therapeutic effect. An alternative strategy to solve this hurdle is gene delivery mediated by an integrative vector.
Using this type of approach, Siew et al. tested PFIC3 correction by the use of an integrative hybrid vector based on the expression of a piggyBac transposase and an AAV8 vector containing a piggyBac ABCB4 expression cassette in FVB
Abcb4-/- mice. A single dose of the hybrid vector in neonates demonstrated the recovery of biliary PC levels and normalization of serum biomarkers. Additionally, the hybrid AAV-piggyBac treatment prevented biliary cirrhosis and reduced tumorigenesis [
116]. However, the possibility of this vector integrating into oncogenic sites represents a high risk for clinical application. Results from these preclinical studies have led to orphan drug designation of an AAV vector harboring a codon optimized version of ABCB4 (VTX-803) developed by Vivet Therapeutics (Paris, France), opening a promising pathway for the treatment of patients with this cholestatic disorder (
Table 2).
Table 2. Gene therapy approaches for PFIC3.
Gene Therapy for PFIC3 Targeting Mechanisms of Disease
Although gene supplementation or correction of the affected gene is the most straightforward gene therapy strategy for PFIC3, several studies have shown that it is also possible to treat this disease by altering the expression of other genes that are involved in this pathology. One example is the delivery of vectors that express genes that contribute to the attenuation of liver fibrosis, such as ACE2 and HNF4A, as described in
Section 3.1.3. In this sense, an AAV8 vector expressing ACE2 was able to reduce liver fibrosis in early- and late-stage FVB
Abcb4-/- mice [
117]. Moreover, hepatocyte-targeted administration of
HNF4A mRNA encapsulated with a biodegradable lipid restored the metabolic activity of hepatocytes in FVB
Abcb4-/- mice, leading to a robust inhibition of fibrogenesis [
100].
A novel approach that could be used to treat cholestatic diseases is based on the regulation of BA synthesis and homeostasis. It has recently been described that Limb expression 1-like protein (LIX1L) is increased in the liver of patients with cholestatic diseases and that the normalization of its expression alleviates cholestatic liver injury in different cholestatic mouse models, including FVB
Abcb4-/- mice. LIX1L regulates the levels of miR-191-3p, a microRNA that downregulates transcription factor liver receptor homolog-1 (LRH-1), thereby inhibiting Cyp7a1 and Cyp8b1 expression, two enzymes required for BA synthesis. Based on these data, Li et al. [
118], recently showed that an AAV vector overexpressing miR-191-3p was able to ameliorate cholestasis in FVB
Abcb4-/- mice by direct repression of LRH-1 expression, thereby reducing de novo BA synthesis [
118]. Another potential target for reducing liver fibrosis through gene therapy of cholestatic disorders is the suppression of the neurokinin 1 receptor (NK1R) axis as well as transforming growth factor-β1 (TGF-β1)/miR-31 signaling. In FVB
Abcb4-/- mice, knock-out of NK1R has been shown to decrease the levels of miR-31 and of proinflammatory molecules such as TFG-β1, resulting in the reduction of liver inflammation and fibrosis [
119]. These therapeutic approaches could be very useful for either acquired cholestatic disorders or PFIC.
Gene Therapy for Other Types of PFIC
For other types of PFIC, although gene supplementation using vectors expressing the specific mutated gene is also an option, there are certain barriers that make the development of these treatments more challenging than for PFIC3. For example, patients with PFIC1, PFIC4, PFIC5, and PFIC6 have extrahepatic manifestations that cannot be rescued by liver-targeted gene therapy [
109,
120]. In addition, in contrast to gene therapy for PFIC3, where liver toxicity arises in the bile canaliculi and transgene delivery to a fraction of hepatocytes leads to sufficient ABCB4 protein to reverse toxicity, in other types of PFIC where toxicity occurs in hepatocytes, it is likely that correction of a high percentage of these cells will be required to achieve a therapeutic effect [
110,
121]. One additional problem to develop gene therapy strategies for some types of PFIC is the lack of suitable animal models that adequately recapitulate the phenotype of patients. Currently, there are no
TJP2-deficient animal models available to test the feasibility of gene therapy for PFIC4 [
121]. Likewise, the existing animal model for PFIC6 is not suitable, because it has a complete knock-out of the MYO5B protein, which is not an appropriate model for this cholestatic disease. For that, it is necessary to develop an animal model with missense mutations of the
MYO5B gene that affect the motor domain but do not result in complete deficiency of the protein [
122]. In the case of PFIC2, there are several animal models that show a varying degree of pathology depending on the genetic background.
Abcb11-/- mice in a C57BL/6 background represent the closest model to the patient disease phenotype, showing a drastic decrease in bile salt content in the bile that leads to increased levels of serum transaminases, liver fibrosis, and hepatomegaly, with these changes being more severe in females than in males [
123]. However, unlike PFIC2 patients, these mice only show a mild elevation of serum bile salts, which is one of the main biomarkers of the disease.
Finally, the loss of transgene expression by hepatocyte cell division is a drawback for the use of non-integrative vectors, such as AAV, in gene therapy of these inherited cholestatic disorders that need to be treated at very early ages, as only a few hepatocytes will maintain episomal AAV genomes [
124]. Unlike PFIC3, for which partial gene therapy supplementation or correction of the affected gene is feasible, other types of PFIC may benefit from other gene therapy strategies aimed at reversing liver damage at several levels.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10061238