Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Flavonoids are widely occurring secondary metabolites of plants. Currently, there is a trend of article numbers increasing, which focuses on the computer modeling of flavonoid interactions with biological targets. Such studies help to accumulatethe data on lead compounds that can find medicinal implementation, including COVID-19. Flavanonol taxifolin demonstrated wound-healing activity. Luteolin, apigenin, and wogonin, which can be classified as flavones, show induced neutrophil apoptosis and have potential as neutrophil apoptosis-inducing anti-inflammatory, proresolution agents.
- flavonoids
- phytomedicine
- taxifolin
- molecular modeling
- COVID-19
1. Structure—Biological Activity Relationship: Qualitative Analysis
The parent structure of flavonoids is 1,3-diphenylpropane, and the aromatic fragments are designated as ring A and ring B [67]. The majority of flavonoid groups are characterized by the heterocycle (ring C) containing oxygen. This ring may be aromatic (flavones, flavonols, etc.) or not (flavanones, flavanonols, etc.). As the rule, carbonyl and several hydroxyl functional groups are present in the molecular structure of flavonoids that can act as a pharmacophores.
The phenolic hydroxyl groups of the studied natural compounds serve as H-bond donors. In cases when the hydrophobic interactions play a key role, the presence of the methoxy group leads to an increase of affinity to the target compared with the hydroxyl group [68]. Due to aromatic rings, the π,π-interactions with the side residues of heterocyclic and aromatic α-amino acids (tryptophan, histidine, phenylalanine, and tyrosine) are possible [69]. Figure 1 demonstrates all types of interactions.
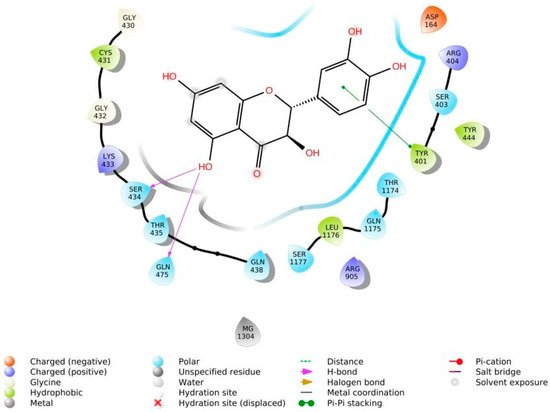
Figure 1. Interaction of taxifolin and P-glycoprotein.
It was found that the antiangiogenic potential of the flavonoid depends on the presence of a C2-C3 double bond [70]; the hydroxyl group in the position 3′ of the ring C contributes to an increase in antioxidant, anti-inflammatory, and antitumor activity [71]. If, along with the multiple C2-C3 bonds, a catechol group is present in the ring B, then such a molecule demonstrates a high affinity for the angiotensin-converting enzyme [72]. Substituents 3-OH, 5-OH, 6-OMe, 6-OH, 7-OH, 3′-OH, and 4′-OMe were identified as key fragments of the molecules when interacting with multidrug resistance-associated protein 2 (MRP2) [55].
It was also interesting to determine the specificity of the interaction of flavonoid groups. Thus, flavones (6-hydroxyluteolin, scutellarein), flavonols (kaempferol), and flavanones (naringenin, eridioctyol) exhibit a high affinity to the estrogen receptor α (ERα), which has been proven in both AutoDock and Glide software. Representatives of these groups of flavonoids can be recommended in the development of antitumor drugs for the treatment of breast cancer [71,73]. Interaction with this protein target results in several types of patient management, such as estrogen hormone replacement therapy and preventive care for breast cancer [74]. Flavones (baicalein, ladanein), flavonols (quercetin), and their glycosylated forms (baicalin) interact with the E protein of various strains of the dengue virus causing fever with a similar name [54]. Such ligands may be used in the treatment of this disease [75]. It is worth noting that the width of the confidence interval of the scoring function calculated for flavones is quite large. This indicates a different degree of protein-ligand binding within this group. Flavones (5-hydroxyflavone) and flavonols (quercetin) have a high affinity for the potassium channel Kir6.1, acting on which some cardiovascular diseases can be treated [56]. Flavones (luteolin, apigenin) can serve as the basis of drugs that control the pathogenicity of Helicobacter pylori due to their ability to bind to one of the main virulence factors of bacteria of this species—vacuolating cytotoxin protein (VacA) [76]. Flavonols (quercetin), their glycosides (avicularin, hyperoside), and flavanonols (taxifolin) with comparable effects function as arginase inhibitors, which is a potential target for the development of new approaches to the treatment of leishmaniasis [77]. Flavan-3-ols (catechin, epicatechin) are characterized by the best values of the scoring function when binding to the CA II-F complex in comparison with flavones, flavanones, and flavanonols and are of interest for the treatment of fluorosis [78]. According to the silico results, flavanones (eriodictyol) and flavanonols (taxifolin) are able to inhibit transcription factors Tec1 and Rfg1 because they can be used in the treatment of infection caused by Candida albicans fungus [79].
2. Structure—Biological Activity Relationship: Quantitative Analysis
Meta-analyses of scoring functions calculated during molecular docking was studied in [54,56,71,73,76,77,78,79]. General information about the average affinities of each flavonoid group to the biological targets is presented in Table 1 and Table 2 for AutoDock and Glide software, respectively.
Table 1. Comparison of the average affinity of flavonoid groups to target proteins in the AutoDock.
| Flavonoid Group | Affinity to the Biological Target, kcal/mol * | ||||
|---|---|---|---|---|---|
| ERα | E Protein DENV2-Thai |
E Protein DENV2-My |
Potassium Channel Kir6.1 | Protein VacA | |
| Flavones | −8.3 ± 0.6 | −7.8 ± 1.3 | −7.5 ± 0.9 | −6.7 ± n/a | −8.5 ± 0.3 |
| Flavonols | −7.9 ± 0.5 | −8.4 ± n/a | −8.6 ± n/a | −8.1 ± n/a | - |
| Flavonol glycosides |
- | −8.1 ± n/a | −7.7 ± n/a | - | - |
| Flavanones | −8.5 ± n/a | - | - | - | - |
| Flavanonols | −9.0 ± n/a | - | - | - | - |
* A lower value of the scoring function corresponds to a better binding.
Table 2. Comparison of the average affinity of flavonoid groups to target proteins in Glide.
| Flavonoid Group | Affinity to the Biological Target, kcal/mol * | ||||
|---|---|---|---|---|---|
| ERα | Complex CA II-F | Arginase | Tec1 | Rfg1 | |
| Flavones | −8.5 ± 0.3 | −3.3 ± 0.0 | - | - | - |
| Flavonols | −8.8 ± n/a | - | −8.1 ± n/a | - | - |
| Flavonol glycosides |
- | - | −8.2 ± 0.3 | - | - |
| Flavanones | −10.2 ± n/a | −2.7 ± 0.2 | - | −7.7 ± n/a | −6.7 ± n/a |
| Flavanonols | - | −2.9 ± n/a | −8.2 ± n/a | −7.7 ± n/a | −4.9 ± n/a |
| Flavan-3-ols | - | −4.7 ± 0.6 | - | - | - |
| Isoflavones | −9.0 ± 0.20 | - | - | - | - |
| Dihydrochalcones | −8.3 ± n/a | - | - | - | - |
* A lower value of the scoring function corresponds to a better binding.
Table 1 provides the experimental data on docking flavonoids of different groups to several biological targets, as performed by AutoDock. The extracted affinity values demonstrate the potential ability of small molecules to form supramolecular complexes with selected proteins in their active sites resulting in multiple pharmacological effects. Apparently, flavanonols form significantly more stable complexes with ERα compared with flavonols, which was approved by in vitro experiment [57]. This may be explained by the non-plane structure of the carbon skeleton, which can be quite similar to estrogen. Furthermore, the affinity value for flavones with protein VacA seems high compared to other targets, so it may be interesting to perform similar calculations for other flavonoid groups.
The results obtained from the meta-analysis of affinity values of flavonoid groups to target proteins calculated via Glide are summarized in Table 2. Compared with the previous table, the heterogeneity of values is obvious. The scoring functions for flavonoids docking with complex CA II-F and RFG1 were notably lower than for other biological targets. Surprisingly, the results of Glide calculations confirmed the previous in silico data obtained by AutoDock: Flavonones demonstrate a significantly higher affinity with ERα and RFG1 than other flavonoid groups. At the same time, flavan-3-ols show significantly higher scoring functions with complex CA II-F.
In general, the data of Glide calculations seemed more appropriate for decision support in further study design due to significant differences in affinity values compared with AutoDock.
3. Lead Compounds
Molecular docking makes it possible to evaluate the affinity of the ligand with the target. Based on this criterion, it is possible to compile a list of the most active compounds for each specific interaction of a flavonoid with a macromolecule. The structures of the leader compounds are shown in Figure 2. Thus, taxifolin (dihydroquercetin) has a high potential for tuberculosis therapy, as it has demonstrated the ability to interact with DNA gyrase and aminoacyl-tRNA synthetase—two enzymes involved in the translation, transcription, and replication of bacterial DNA [97]. The pathogenic effect of the Ebola virus can be disrupted by affecting the vital protein structures VP40, VP35, VP30, and VP24 with flavonoids such as gossypetin or taxifolin. These compounds proved to be leaders in the study of more than 4500 flavonoids [98]. It is interesting to notice that despite the near structures of the molecules, they have similar interaction patterns with only two proteins among four targets. Both taxifolin and gossypetin can form H-bonds with His124, Gly126, and Gln170 in VP40. Furthermore, they interact with Gln103, Ser123, Asp124, and Asn185 or Gln184 in the active site of VP24. Key amino acid residues in VP35 are Gln244(2) and Asp302 for taxifolin, while for gossypetin, it is only the Gln241(2). Finally, gossypetin interacts with Asp158(2), Arg196, and Gln233(2) in VP30. However, taxifolin forms H-bonds with Arg196, Gly200(3), Gln233, Ser234, and Phe238 residues of the VP30. Differences in the affinity of these flavonoids are apparently associated with the spatial structure of the heterocyclic ring. Gossypetin has a plane aromatic structure, and the taxifolin molecule is characterized by two chiral centers. Thus, taxifolin can exist as four stereoisomers. Saltillin, taxifolin, and 6-methoxyflavone have a high affinity for N-myristoyltransferase (NMT), a target for the treatment of candidiasis [99]. There is also information indicating the feasibility of studying a number of flavonoids in the following diseases: lung cancer [100], breast cancer [50,71], metabolic syndrome [49], pathological conditions caused by Pseudomonas aeruginosa [101], hypoestrogenism [102], and depressive disorders [103].
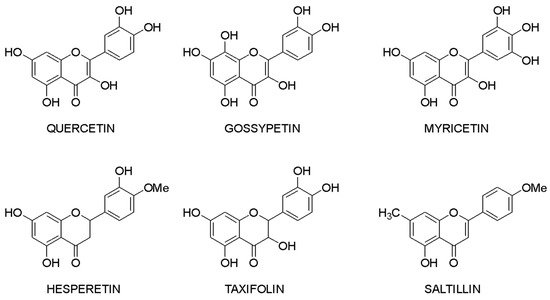
Figure 2. Lead compounds.
Taxifolin is one of the most promising lead compounds, mentioned in Figure 2, due to its wide range of pharmacological activity, rich raw material base, and high safety profile. Prospective taxifolin use in medicine was discussed in a number of articles and reviews [104,105,106]. Moreover, in Russia, this compound is registered as an active pharmaceutical ingredient, and in Europe, it is a supplement (Regulation EU 2017/2470). (2R,3R)-taxifolin is the most investigated isomer.
It is known that the (2R,3R)-taxifolin obtained from natural sources is safe [107]. The low toxic potential of taxifolin was predicted by different methods, such as the ORISIS DataWarrior program [108]. Furthermore, it is quite important to note that not only the major trans-stereoisomer is characterized by a high safety profile. The VirtualToxLab platform determined (2S,3S)-taxifolin as one of the lowest toxic molecules from the set of 29 molecules that demonstrated affinity to the SARS-CoV-2 main protease [109]. This software is based on calculating individual binding affinities to 16 validated off-targets, including intracellular receptors, metabolic enzymes, and the hERG potassium channel. Taxifolin is the safest flavonoid in comparison with eriodictyol, luteolin, isoscutellarein, and quercetin. It has shown in silico potential as the main protease of SARS-CoV-2 [108].
Despite the great pharmacological potential, this flavonoid is characterized by low bioavailability [110,111]. To explain the observed efficacy, research on taxifolin metabolism was performed. More than 190 structures of different compounds were found as the result of taxifolin biotransformation by the HPLC-MS/MS method [112]. There was research that tested most of them as COX-2 inhibitors by in silico study [39]. To evaluate the affinity of metabolites to target, molecular docking was performed. A significant number of these compounds were absent in databases. For this reason, the ligand set of 214 3D-structures of all taxifolin stereoisomers and their metabolites was generated manually using ChemBioDraw Ultra (v. 13.0, PerkinElmer, Waltham, MA, USA). Then, the GOLD software (v. 5.4, CCDC, Cambridge, UK) was used to conduct the modeling of intramolecular interaction. During this research, all metabolites were classified into three groups. Compounds that contain all three rings of the initial structure can interact with the three most important amino acid residues in the active site of the target: SER353, SER530, and ARG513. Metabolites that contain two rings (A, C or B, C) do not interact with ARG513 and, for this reason, are less selective. The third group consists of molecules with only one ring. These compounds have a low affinity to COX-2. Thus, research in the field of the design of anti-inflammatory drugs based on taxifolin should be continued.
Crystal engineering is another area of taxifolin research. Microtubes are one of the most significant achievements in the development of new crystal forms in the last 5 years [113]. They were synthesized by precipitation with water from an ethanol solution of taxifolin in the presence of urea. This modification showed notable differences in comparison with the initial active pharmaceutical ingredient. To characterize the properties of the new taxifolin forms in silico, the 3D-models of nanoparticles were generated based on the different crystal unit structures. The simulation of the nanoparticle deformation was carried out by molecular dynamics modeling to evaluate the physical characteristics of taxifolin modifications. This investigation was performed using Materials Science Suite 2018-2 (Schrodinger, New York, NY, USA). A cross-shaped pore was observed at the core of the taxifolin nanoparticle (Figure 3). The diameter of this pore varied from 4 nm at the narrow part to 11 nm at the widest part. These findings make it possible to suggest that the taxifolin microtubes are a mesoporous material. Taxifolin microtubes may have an application in drug delivery.
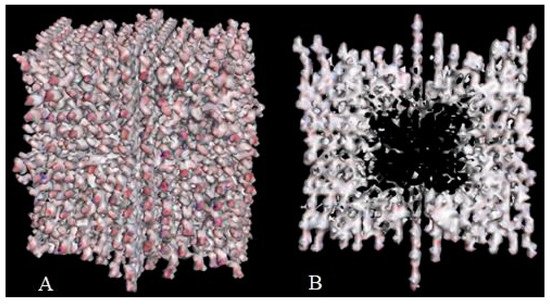
Figure 3. The view of taxifolin nanoparticle (A) and its cross-section (B) [113].
A wide range of pharmacological activity of flavonoids is a potential for the design of drugs on these bases. After the in silico stage, preclinical and clinical trials are required to verify the safety and efficacy of the candidate molecule. At this moment, on the website https://clinicaltrials.gov (accessed on 25 January 2021) 253 clinical trials of drugs containing flavonoids have been registered. It is worth noting that the number of trials currently being conducted or planned for the near future is 45, and these include pathologies such as post-thrombotic syndrome, sickle cell anemia, chronic kidney disease, psoriasis, glaucoma, type 2 diabetes, and others. It is interesting to note that the possibility of flavonoids being used for the COVID-19 treatment is being studied in 11 trials.
4. COVID-19 Treatment
Undoubtedly, the Coronavirus disease 2019 (COVID-19) pandemic is one of the most socially important problems nowadays. There have been approximately 448 million confirmed cases of COVID-19, including more than 6 million deaths, reported by the World Health Organization (WHO) [114]. According to these epidemiological data, it is extremely important to find effective remedies against COVID-19. The flavonoid group is a promising object for this purpose. Indeed, thousands of investigations have been carried out. Obviously, most of them were conducted by in silico approaches.
For instance, one study aimed to establish flavonoid’s affinity to the spike protein of SARS-CoV-2. The ligand set included apigenin, chrysin, fisetin, galangin, hesperetin, luteolin, morin, naringin, quercetin, and rutin. Docking studies showed that all flavonoids demonstrate considerably high binding affinity, but naringin is characterized by the highest one. This compound shared hydrophobic interactions with the following residues: Asn290, Ile291, His374, Leu370, Leu410, Ala413, Pro415, Phe438, and Gln442. Furthermore, hydrogen bonds were formed with Asp367, Thr371, Lys441, Glu406, and Ser409 amino acid residues [115].
Six polyphenolic compounds, namely leucopelargonidin, morin, myricetin, eriodictyol, taxifolin, and enterodiol, exhibited a significant binding affinity for SARS-CoV-2 papain-like protease (PLpro) and main protease (Mpro) during another investigation via molecular docking [116]. The analysis of free binding energy showed that taxifolin has the highest affinity against Mpro and forms H-bonds with Cys145, Ser144, Gly143, Asn142, Leu141 (hydroxyl groups of ring A), Glu166, Met165, His164 (carbonyl group of ring C), Tyr54, Pro52, and Met49. Morin was determined as a compound with the highest affinity toward Plpro. Hydrogen bonding interactions of morin were observed with the following amino acid residues: Gly266, Asn267 (hydroxyl groups of rings A and B), Thr301 (carbonyl group of ring C), Tyr273, Tyr264, and Tyr268 (phenolic hydroxyl groups of ring B). Moreover, further molecular dynamics simulation showed that the binding of bioactive molecules on the corresponding proteases is characterized by structural changes that can disrupt the functions of enzymes and, thus, enhance their antiviral activity.
Another approach considers angiotensin-converting enzyme 2 (ACE-2) receptors as a target. Molecular docking was used to predict the activity of flavonoid sets, including hesperetin, chrysin, kaempferol, galangyn, myricetin, and rutin [117]. The last molecule showed the best binding affinity to the ACE-2 enzyme. It was found that rutin forms the strongest hydrogen bond with Asn149, Met270, His345, Lys363, Asp368, and Thr445 amino acid residues of the ACE-2 protein. Likewise, this compound has the π-anion interaction with Arg269, π-π stacking interaction with Phe274, π-alkyl interaction with Ala153, and alkyl interaction with Pro346, Met360, and Cys361 residues.
Moreover, in our recent study, 163 flavonoids were screened [118]. ATP-binding domain SP3, main protease NSP 5, RNA-dependent RNA polymerase NCP12, and endoribonuclease NCP15 were considered as targets. Much of the recent COVID-19 treatment research in silico has focused on the identification of lead compounds, while our study pays more attention to the binding affinity of the all-flavonoids groups to biological targets. The median binding energies were −7.4, −7.4, −8.9, and −7.3 kcal/mol for the ADP-binding domain NSP3, main protease NSP5, RNA-dependent RNA polymerase NSP12, and endoribonuclease NSP15, respectively. Therefore, these data suggest that flavonoids can find application in the complex therapy of COVID-19.
The potential benefits of flavonoids in COVID-19 therapy were confirmed by a randomized controlled trial [119]. Groups that obtained quercetin in combination with antiviral drugs demonstrated significantly lower serum levels of critical markers involved in COVID-19 severity and better respiratory rate. However, further clinical trials with other flavonoids and bigger group sizes are required.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23116023
This entry is offline, you can click here to edit this entry!