Interestingly, members of the single-subunit (parkin, SIAH, CHIP) and multiple-subunit (SCFFXBL5) RING domain E3 ligase family, as well as the HECT domain family of E3 ligases (NEDD4 family), have been found capable of ubiquitinating α-synuclein.
The precise cell type-specific expression of NEDD4-1 and NEDD4-2 in the midbrain dopaminergic system requires further analysis. However, their expression in midbrain dopaminergic neurons not only during development but during adulthood and ageing, as well as in PD brain samples, supports the idea of an important role here for protein homeostasis.
Alterations in the phosphorylation of NEDD4-1 and NEDD4-2 have been widely observed to regulate their ubiquitination activity and alter the binding of adaptor or scaffold proteins. Fibroblast growth factor receptor 1 (FGFR1) is a substrate of NEDD4-1 ubiquitination that triggers c-Src kinase-dependent phosphorylation of NEDD4-1 at Tyr
43 in the C2 domain and Tyr
585 in the HECT domain, supporting activation [
43]. For NEDD4-2, G-protein-coupled receptor (GPCR) protease-activated receptor-1 (PAR1) stimulates c-Src-mediated Tyr
485 phosphorylation within the 2,3-linker peptide between WW domains 2 and 3 and leads to NEDD4-2 activation [
227]. Phosphorylation of
Xenopus NEDD4-2 on Ser
338 or Ser
444 by the serine/threonine kinase serum- and glucocorticoid-induced kinase 1 (SGK1) was shown to lead to a reduction in its affinity for the natural NEDD4 substrate epithelia Na
+ channel (ENaC), which regulates whole-body Na
+ balance and blood pressure [
228,
229]. Human NEDD4-2 phosphorylation by aldosterone-induced SGK1 on Ser
342 and Ser
448 (and Thr
367) was shown to facilitate 14-3-3 protein binding to NEDD4-2, leading to inhibition of the interaction between NEDD4-2’s HECT and WW domains, stabilisation of ENaC in the kidney, and enhanced ubiquitination of the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor subunit GluA1 (glutamate ionotropic receptor AMPA type subunit 1) in the brain. For
Xenopus NEDD4-2, it was suggested that the 14-3-3 dimer binds first on NEDD4-2 P-Ser
444, the high-affinity (major) site, and subsequently on one of the lower-affinity (minor) sites, P-Ser
338 or P-Thr
363 [
230]. For human NEDD4-2 it has been shown that the 14-3-3 dimer simultaneously anchors on two of the three phosphorylation sites, P-Ser
342, P-Thr
367 and P-Ser
448, of NEDD4-2, with P-Ser
448 being the key residue [
93]. SGK1 also leads to phosphorylation of human NEDD4-2 Ser
468 and an increase in ENaC protein [
92,
231]. Interestingly, SGK1 has also been suggested to be a NEDD4-2 substrate, leading to its own degradation and generating a negative feedback loop [
232]. The same three SGK1 phosphorylation sites, Ser
342, Ser
448 and Thr
267, of NEDD4-2 are also used by vasopressin-induced cyclic AMP-dependent protein kinase A (PKA). Furthermore, insulin activates SGK1 and Akt (protein kinase B) and leads to Ser
342 and Ser
428 phosphorylation of human NEDD4-2, upregulating ENaC on the membrane [
155]. IkappaB kinase β (IKKβ) has been found not only to bind to ENaC and enhance its activity but to phosphorylate
Xenopus NEDD4-2 on Ser
444, preventing NEDD4-2-dependent ENaC ubiquitination [
117]. Interestingly, 14-3-3η has also been shown to bind and inhibit the ubiquitination activity of wildtype parkin but not of parkin with R42P, K161N, and T240R mutations associated with autosomal recessive juvenile parkinsonism [
233]. The parkin/14-3-3 inhibitory complex could be prevented by wildtype α-synuclein but not by A30P and A53T mutations, causing PD [
233]. These data define the chaperone-like protein 14-3-3 as an important inhibitor of E3 ligases associated with PD.
The regulatory mechanisms occurring in the midbrain dopaminergic system to change the activity or substrate specificity of NEDD4-1 and NEDD4-2 have so far not been investigated but are likely to utilise at least some of the aforementioned post-translational modifications and interaction partners. With further information about important in vivo substrates and functions of NEDD4-1 and NEDD4-2 in dopaminergic neurons, it will be a revealing task to study the detailed regulatory mechanisms.
5. NEDD4-1 and NEDD4-2 Substrates, Adaptors, Regulators, and Function
Upon the discovery of NEDD4-2, it was proposed that NEDD4-1 and NEDD4-2 may have redundant functions with shared interaction partners and substrates, however there appears to be adaptors, substrates, and functions specific or unique to NEDD4-1 and NEDD4-2.
The different phenotypes of NEDD4-1 and NEDD4-2 knockout (KO) mice suggest that their main substrates are distinct, and the redundancy might be limited to a few substrates and functions [
42]. The predominant phenotype of NEDD4-1 KO mice is embryonic lethality at midgestation, with pronounced heart defects (double-outlet right ventricle and atrioventricular cushion defects) and vasculature abnormalities leading to growth retardation (with a body weight less than 40% of that of wild-type littermates) [
46,
117,
118]. In contrast, NEDD4-2 KO mice show perinatal lethality, with increased ENaC levels that seem to cause premature foetal lung fluid clearance, resulting in a failure to inflate the lungs [
198]. Only a few of these mice survived up to 22 days [
198]. This phenotype was also confirmed in lung-specific NEDD4-2 deficient mice [
234]. When crossing floxed NEDD4-2 mice with EIIa-Cre mice (B6.FVB-Tg(EIIa-cre)C5379Lmgd/J) [
235] expressing Cre in a mosaic pattern in the embryo before implantation in the uterine wall, the NEDD4-2 KO mice might not be a complete null for NEDD4-2 [
236]. These mice were viable but showed defects in the respiratory, renal, cardiac, neural, and immune systems and high blood pressure, indicating that NEDD4-2 is a key regulator of Na
+ homeostasis and that ENaC is one of its most important physiological substrates [
236]. This suggests that even ENaC can be a substrate of NEDD4-1 and NEDD4-2 [
42]. The NEDD4-1 and NEDD4-2 KO mouse data suggested that in vivo NEDD4-2 is most likely the more important E3 ubiquitin ligase for ENaC. Interestingly, the G protein-coupled receptor kinase 2 (Grk2) can phosphorylate ENaC on Ser
633 in the C-terminus of the β-subunit, which increases ENaC activity and prevents ENaC ubiquitination by NEDD4 ligases and subsequent degradation [
237]. GRK2 and other GRK family members have also been described to phosphorylate α-synuclein on Serine
129, which is common in PD patients, however it has not been reported whether this phosphorylation negatively influences ubiquitination by NEDD4 ligases [
238].
This suggests that NEDD4-1 and NEDD4-2 may have common but also unique functions, and this might depend on the specific tissue investigated. Therefore, it seems important to investigate how far NEDD4-1 and NEDD4-2 have redundant and unique functions in vivo during the development and maintenance of the midbrain dopaminergic system as well as in pathophysiological conditions leading to PD.
Most NEDD4-1 and NEDD4-2 substrates and adaptors have so far been investigated only in vitro and await in vivo confirmation. PTEN (phosphatase and tension homologue) is a good example to illustrate the importance of verifying possible NEDD4 substrates in vivo under physiological conditions in an organism such as the mouse. Cell culture experiments and human cancer tissue suggested that PTEN might be a NEDD4-1 and NEDD4-2 substrate [
177,
178,
179,
180,
239]), but analysis of NEDD4-1 and NEDD4-2 knockout mice has shown that PTEN stability, subcellular localisation, and activity are not altered in the absence of NEDD4-1 and/or NEDD4-2 [
181,
182]. Furthermore, more recent cell culture experiments have not supported PTEN as a NEDD4 substrate [
225]. Therefore, more research would be required to finally resolve this controversy between in vitro and in vivo data.
The adaptor proteins NDFIP1 (NEDD4 family-interacting protein 1 or NEDD4 WW domain-binding protein 5 (N4WBP5)) and NDFIP2 (NEDD4 family-interacting protein 2 or NEDD4 WW domain-binding protein 5A (N4WBP5A)) are small, endosomal, PY-motif-containing membrane proteins that can both function as adaptors for NEDD4-1, NEDD4-2, ITCH/AIP4, WWP1, and WWP2, facilitating their binding to proteins that lack PY motifs, preventing autoinhibition of the ligase, and possibly serving as ubiquitination substrates. Overexpression of NDFIP1 is able to recruit NEDD4-1, NEDD4-2, and ITCH to neuronal exosomes, which are normally free of these E3 ligases, for secretion [
109,
203]. The positive effect of NDFIP1/NEDD4-1 in improving neuronal survival during brain injury suggests that perhaps exosomal NEDD4-1 might enhance transport and degradation of unwanted proteins [
203]. As microglial exosomes facilitate the transmission of α-synuclein in PD [
240], it would be of interest to study the role of NEDD4 ligases in this process. NDFIP1 and NDFIP2 are physically and functionally associated with multiple components of the epidermal growth factor (EGF) signalling cascade, and their levels modulate the relative output of different signalling pathways. They associate with the EGF receptor and the phosphatase and tension homologue (PTEN) and control the ubiquitination and abundance of PTEN, cellular Casitas B-linage Lymphoma E3 ligase (c-CBL), and cellular Sarcoma family kinases (c-Src). NDFIP2, but not NDFIP1, also binds to and is phosphorylated by two c-Src kinases (Src and Lyn) and can act as a scaffold for Src phosphorylation of NDFIP1 and potentially other substrates. Depletion of NDFIP1 inhibits serine/threonine kinase Akt (protein kinase B, PKB) activation in EGF-stimulated HeLa cells, stimulates activation of cellular transcription factor c-Jun-N-terminal Kinase (Jnk), and enhances cell multiplication. Interestingly, increased iron is often found in the substantia nigra of PD patients and has been associated with increased NDFIP1 levels [
239]. It would be of interest to examine whether iron misregulation may serve to be protective to nigral dopaminergic neurons by upregulating NDFIP1 and facilitating NEDD4-1-mediated ubiquitination of α-synuclein.
Adaptor proteins such as NDFIP1 and NDFIP2 seem to use different members of the NEDD4 family in vivo. NDFIP1-deficient mice showed a reduced life expectancy, with severe inflammation of the skin and lung, enhanced T-cell activation, proliferation and differentiation to T helper 2 cells, and a prolonged JunB half-life such as that in in Itchy mutant mice lacking functional ITCH protein [
192,
241]. NDFIP2-deficient mice showed no overt immunopathology, but NDFIP2 deficiency seemed to enhance the NDFIP1 knockout phenotype, leading to further accumulation of effector CD4+ T cells and an increase in JAK (Janus kinase) protein, which might be explained by reduced Itch or NEDD4-2 activation [
242]. Further research has to be done to confirm the use of the adaptor proteins NDFIP1 and NDFIP2 by NEDD4-1 and NEDD4-2.
NEDD4-1 activation has also been shown to be important for autophagy and mitophagy [
66,
243]. LC3 (MAP1LC3, microtubule-associated protein 1 light chain 3) is essential in autophagy by functioning in elongation of the phagophore double-layer membrane and in the recruitment of proteins for autophagic processes. LC3 activates and recruits NEDD4-1 to the phagophore assembly site (PAS) by binding the conserved WXXL LC3-binding motive between the C2 and the WW2 domains. LC3-I is activated to LC3-II by cleavage and conjugation to phosphatidylethanolamine (PE) and is recruited to autophagosomes by binding LIR (LC-3 interacting region domain-containing protein). Subsequently, NEDD4-1 ubiquitinates the LC3-interacting protein p62 (sequestosome-1, SQSTM1) and beclin-1 (BECN1), which seems required to recruit downstream effectors for autophagosome formation [
32,
66]. More recently, NEDD4-1 lysine
29-linked autoubiquitination on lysine
1279 was shown to recruit USP13 (ubiquitin-specific protease 13) to form a deubiquitination complex, which stabilised VPS34 to promote autophagy by removing the lysine
48-linked polyubiquitin chains from VPS34 at lysine
419 [
74]. Surprisingly, in mice, endoplasmic reticulum (ER) stress and activation of the unfolded protein response (UPR) increased autophagy and NEDD4-2 expression in the liver, but not NEDD4-1 expression. In addition, in cell culture, high amounts of NEDD4-2 correlated with increased autophagy, while low amounts of NEDD4-2 correlated with reduced autophagy [
244]. In PD, reduced autophagy is a common phenotype that can be triggered by α-synuclein accumulation and might be enhanced by NEDD4-1 and NEDD4-2 [
245].
Interestingly, NEDD4 ubiquitination activity is required for the release of some retroviruses but might be inhibited as a cellular defence mechanism. The Gag protein of human oncoretrovirus HTLV-1 (human T-lymphotropic virus type 1) has a tandem PPPY/PTAP motif and needs to be ubiquitinated by the E3 ligase NEDD4-1 at the plasma membrane. It also requires Tsg101 (tumour susceptibility gene 101) recruitment at the ESCRT (endosomal sorting complexes required for transport) pathway in late endosomes/multivesicular bodies for driving virus budding [
49]. Despite the HIV (human immunodeficiency virus) Gag protein lacking a PY motif, it also uses NEDD4-1 and NEDD4-2 for its ubiquitination, to stimulate budding; Nedd4-2, with the adaptor protein AMOT-1 (angiomotin-1 protein); and NEDD4-1, by binding and ubiquitinating adaptor protein ALIX (apoptosis-linked gene 2 (ALG-2)-interacting protein X, programmed cell death 6-interacting protein) [
246]. However, NEDD4 family members might be inhibited in cells after viral or bacterial infection by binding with upregulated interferon-induced ubiquitin-like protein ISG15 (interferon-stimulated gene 15). The binding of ISG15 to NEDD4-1, NEDL1, NEDL2, or WWP2 can block their interaction with ubiquitin-E2 enzymes and interfere with the ubiquitination of retroviral group-specific antigen precursors and matrix proteins, such as VP40 of Ebola with a PPxY motif, which is essential for the release/budding of Ebola, vesicular stomatitis, and rabies virus particles [
247,
248]. DNA virus proteins such as the latent membrane protein 2A (LMP2A) of Epstein–Barr Virus (EBV, human herpesvirus 4) also interact via their PPPPY motif with the NEDD4 family members NEDD4-1, ITCH, and WWP2 [
68]. This interaction leads to the ubiquitination of LMP2A and LMP2A-associated proteins such as the protein tyrosine kinases Lyn (Lck/Yes novel tyrosine kinase of the Src kinase family) and Syk (spleen tyrosine kinase), which might be important for EBV latency and the regulation of B-cell signal transduction [
68]. Taken together, this suggests an important role of NEDD4 family members in vesicular transport and retrovirus propagation being regulated by the immune system, which have been found to be altered also in neurodegenerative diseases such as PD [
249].
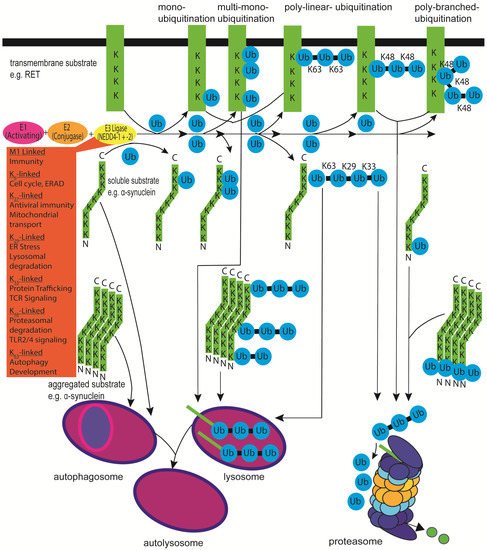
In the context of PD, most research in the past focused on NEDD4-1 after NEDD4-1 was found to ubiquitinate α-synuclein as detailed above [
28,
29]. An interesting NEDD4-1 substrate in this regard is the receptor tyrosine (and serine) kinase RET (abbreviation for rearranged during transfection), the canonical receptor for the TGF-β (transforming growth factor-β)-related neurotrophic factor family member GDNF (glial cell line-derived neurotrophic factor), which is currently in clinical trials in PD patients [
245,
250,
251,
252] (see
Figure 1). The RET receptor is important for the maintenance, protection, and regeneration of midbrain dopaminergic neurons [
253,
254,
255]. In cell culture experiments, the turnover of the long splice isoform of RET, RET51, is mediated after activation and autophosphorylation by binding of the adaptor protein GRB2 (growth factor receptor-bound protein 2) to RET tyrosine
1096 and subsequent recruitment of the E3 ubiquitin–protein ligase c-CBL [
82]. However, the short RET isoform, RET9, binds to the adaptor proteins GRB10 or SHANK2 (SH3 And Multiple Ankyrin Repeat Domains 2). This depends on its phosphorylated tyrosine
1062 and the C-terminal PDZ-binding motif (PDZ is an initialism combining the first letters of the first three proteins discovered to share the domain—postsynaptic density protein (PSD95), Drosophila disc large tumour suppressor (Dlg1), and zonula occludens-1 protein (zo-1)) and recruits NEDD4-1 [
82]. RET polyubiquitination triggers receptor internalisation from clathrin-coated pits at the cell membrane into endosomal compartments for receptor recycling to the cell surface or lysosomal degradation [
256]. RET51 shows more K63 ubiquitin linkage—in contrast to RET9—and can be sorted to RAB11-positive recycling endosomes for signalling, intracellular trafficking, and return to the cell surface or targeted for lysosomal degradation [
257,
258]. RET9 ubiquitination chains are more K48 linked, targeting the protein more for proteasomal degradation [
259]. Determining whether NEDD4-1-dependent ubiquitination of RET9 also occurs in dopaminergic neurons in vivo and influences survival and physiology requires further investigation.
Other receptor tyrosine kinases have also been suggested to be substrates of NEDD4-1, such as the fibroblast growth factor receptor 1 (FGFR1) [
43] and the epidermal growth factor receptor (EGFR) members ErbB1 (erythroblastic leukaemia viral oncogene homologous-B2 receptor tyrosine kinase 1) [
225] and HER3/ErbB3 (human epidermal growth factor receptor 3) [
53]. NEDD4-1 also mediates the adaptor protein β-arrestin2’s agonist-dependent ubiquitination and lysosomal degradation of the β2-adrenergic receptor (β2AR) [
33]. IGF1R (insulin-like growth factor I receptor) can bind the adaptor protein GRB10, and this was suggested to mark IGF1R for NEDD4-1-dependent polyubiquitination and degradation [
168,
260,
261] or protect IGF1R from NEDD4-1 ubiquitination [
262]. Further work is needed to understand this controversial NEDD4-1 and IGF1R crosstalk. VEGFR-2 (vascular endothelial growth factors receptor 2), but not VEGFR-1, has also been suggested to be protected from NEDD4-1-induced degradation by binding GRB10, although VEGFR-2 might not be a direct NEDD4-1 ubiquitination substrate [
91]. VEGF stimulation of VEGFR-2 increases GRB10 expression and c-Src-dependent tyrosine phosphorylation of GRB10, which subsequently increases VEGFR-2 protein levels [
263]. Interestingly, in NEDD4-1 knockout mice, GRB10 protein levels were increased and IGF1 and insulin signalling were reduced, while
GRB10 gene deletion rescued the NEDD4-1 knockout lethality, suggesting a negative regulatory function of GRB10 for IGF1 and insulin signalling [
262]. These data suggest that NEDD4-1 might not only directly targets receptor tyrosine kinases as substrates but also indirectly regulate receptor tyrosine kinase signalling by targeting associated adaptor proteins. For example, NEDD4-1 monoubiquitinates insulin receptor substrate (IRS)-2, which promotes its binding to the clathrin-coated pit adaptor protein epsin-1 and the recruitment of IGF1R, which phosphorylates IRS-2, stimulating downstream signalling [
59]. Another NEDD4-1 monoubiquitination substrate seems to be the adaptor protein HGS (hepatocyte growth factor-related kinase substrate, HRS), which leads to intramolecular binding of ubiquitin to the HGS ubiquitin-interaction domain (UIM), leading in turn to reduced endocytosis of EGFR [
55]. The secretory carrier membrane protein-3 (SCAMP3) has a PY motif and seems to be multimonoubiquitinated by NEDD4-1, which allows HGS interaction and prevents EGFR degradation [
87]. The endosomal sorting complexes (ESCRT-0, -I, -II, -III, VPS4-VTA1 (vacuolar protein sorting protein 4 and vesicle trafficking protein 1), and ALIX (apoptosis-linked gene 2-interacting protein X) homodimer) are peripheral membrane protein complexes required together for degradation of damaged or unwanted plasma membrane and cytosolic proteins, lysosome and multivesicular body (MVB) biogenesis, autophagy, and viral budding [
264,
265] The ESCRT-0 complex sorts ubiquitinated membrane proteins into MVB. It consists of HGS and STAM (signal transduction adaptor molecules STAM1 and STAM2) proteins and can be associated with the ubiquitin-binding domain-containing protein EPS15B (epidermal growth factor receptor pathway substrate 15B) to mediate EGFR degradation [
265,
266]. EPS15 is associated with clathrin-coated pit adaptor protein 2 (AP2) and plays a role in EGFR internalisation [
266]. The NEDD4 family members NEDD4-1 and ITCH/AIP4 have been found associated with ESCRT complexes, which seems important not only for viral GAG protein ubiquitination and budding but for degradation of a membrane-associated pool of Lys
63 polyubiquitinated α-synuclein [
28,
265]. ESCRT proteins (VPS4 from ESCRT-0, charge multivesicular body protein 2B (CHMP2B) from ESCRT-III) have been found to be important for lysosomal targeting of α-synuclein and are also localised to Lewy bodies [
29,
267,
268,
269]
Both NEDD4-1 and NEDD4-2 can bind the PY motif containing non-receptor tyrosine and serine/threonine kinase ACK (activated Cdc42-associated tyrosine kinase, TNK2). NEDD4-1 leads to polyubiquitination and subsequent lysosomal degradation of ACK along with EGFR in response to EGF stimulation, while the data for NEDD4-2 in regard to ACK ubiquitination remain inconsistent [
151,
152]. Interestingly, FGFR3 activation leads to NEDD4-1 phosphorylation, which subsequently targets the transmembrane protein programmed death-ligand 1 (PD-L1, cluster of differentiation 274 (CD274), B7 homologue 1 (B7-H1)), which is involved in immune system suppression for Lys
48-linked polyubiquitination and degradation [
270]. Furthermore, activation of the small GTPase RAS (rat sarcoma virus protein) is reduced by NEDD4-1 ubiquitinating the PY motif containing the RAS activator CNrasGEF (cyclic nucleotide RAS guanine-nucleotide exchange factor) for proteasomal degradation [
40].
A receptor tyrosine kinase that is suggested to be a NEDD4-2 substrate is the NGF (nerve growth factor) receptor TRKA (tropomyosin receptor kinase A; NTRK1, neurotrophic receptor tyrosine kinase 1) with a PPXY motif, which is, in its activated/phosphorylated state, marked for degradation by NEDD4-2-dependent ubiquitination and is more abundant in the dorsal root ganglia of NEDD4-2 deficient mice [
145,
271,
272]. However, the closely related BDNF (brain-derived neurotrophic receptor) receptor TRKB lacks a PPXY motif and seems not to be a NEDD4-2 substrate [
145]. TRKA is normally not expressed in midbrain dopaminergic neurons, but the ectopic expression of TRKA in vivo in mice combined with NGF treatment protected dopaminergic neurons from 6-OHDA-induced cell death [
273]. TRKB is found in midbrain dopaminergic neurons but is not essential for development and maintenance [
253]. However, TRKB seems to protect cells from MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/1-methyl-4-phenylpyridinium) toxicity in mice [
274].
NEDD4-2 and other NEDD4 family members have also been found to regulate the signalling of the transforming growth factor beta (TGF-β) family of ligands (33 human genes encoding, for example, TGF-β 1, 2, and 3; activins; inhibins; and bone morphogenetic protein (BMPs)). TGF-β family members activate the TGF-β receptor by inducing the heterotetramerisation of the two single transmembrane serine–threonine (and tyrosine) kinase receptors, TGF-β receptors type I and type II [
275,
276]. TGF-β receptor signalling leads to transcriptional regulation of target genes through the canonical SMAD signalling pathway (the SMAD abbreviation refers to the homologies to the
Caenorhabditis elegans SMA (“small” worm phenotype) and the MAD family (“Mothers Against Decapentaplegic”) of genes in
Drosophila) protein involving signalling pathway and the non-canonical (SMAD independent, involving tyrosine-autophosphorylation) [
275,
276]. TGF-β receptor type I has also been described as a NEDD4-2, SMURF1, SMURF2, and WWP1 substrate in conjunction with the inhibitory SMAD7 protein as an adaptor. It is marked for degradation by ubiquitination. The TGF-β I and II receptors signal together through the receptor-regulated SMADs (R-SMADs 1, 2, 3, 5, and 8/9), which can partner with SMAD4 (Co-SMAD) or be suppressed by the inhibitory SMADs (I-SMAD 6 and 7) or the SMAD corepressor SnoN (Ski-relate novel protein N) to downregulate TGF-β receptor signalling [
140,
276]. Interestingly NEDD4-2 was able to bind to all SMAD proteins with a PY motif (1,2,3,5,6,7), but not to those without one (4 and 8), and it induced ubiquitination and subsequent degradation of SMAD2 but not of SMAD3 [
140]. Although NEDD4-2 and SMURF2 can both bind to SnoN via SMAD2, 3, or 4, only SMURF2 can ubiquitinate SnoN and lead to its degradation [
140]. The TGF-β I and II receptors are expressed in dopaminergic neurons, and TGF-β II-deficient mice showed reduced TGF-β I receptor levels, reduced dendritic growth and spine formation, a decreased range of excitatory-to-inhibitory synapses, a reduced excitation/inhibitory ratio (ratio of evoked miniature excitatory postsynaptic currents (mEPSC) to miniature inhibitory postsynaptic currents ratio (MIPSC)), hyperactivity, and a reversal-learning defect but no change in dopaminergic cell counts [
277].
As for α-synuclein, several different ubiquitin–protein ligases have been proposed to influence internalisation, signalling and degradation of most receptor and cytosolic tyrosine and serine–threonine kinases. Therefore, very careful in vivo analysis is needed to understand the roles of NEDD4-1 and NEDD4-2 in regulating a specific kinase, considering in detail the context, such as the organism, developmental stage and age, tissue, and environmental and physiological challenges.
Concerning PD and the function of the dopaminergic system, it is also important to mention that NEDD4-2 can ubiquitinate neurotransmitter transporters. The dopamine transporter (DAT) is required for the reuptake of dopamine into dopaminergic neurons and has been suggested to be ubiquitinated by NEDD4-2, but not NEDD4-1, leading to endocytosis by binding to epsin and Eps15 on clathrin-coated pits [
108,
278]. NEDD4-2 seems to cooperate with the E2 enzymes UBE2D and UBE2L3 to conjugate, in a PKC-dependent reaction, primarily lysine
63-linked ubiquitin chains onto DAT [
108]. Reduced DAT levels increase the amount of extracellular dopamine and prolong the stimulation of pre- and postsynaptic dopamine receptors, leading in mice to locomotion defects [
279]. Another NEDD4-2 substrate that is ubiquitinated in an PKC-dependent manner and reduced in neurodegenerative diseases is the glutamate transporter-1 (GLT-1), which is subsequently internalised and degraded [
280]. In MPP
+-treated astrocytes and MPTP-treated mice, NEDD4-2 mediated the ubiquitination of GLT-1 [
114]. Conversely, NEDD4-2 knockdown increased glutamate transporter protein levels. In MPTP-treated mice, NEDD4-2 knockdown ameliorated movement disorders, increased tyrosine hydroxylase expression in the midbrain, and attenuated astrogliosis and reactive microgliosis associated with glutamate excitotoxicity [
114]. To support the idea that NEDD4-2 might be a therapeutic target for the treatment of PD, it would be of interest to confirm these results in conditional NEDD4-2 KO mice treated with MPTP or a more physiological challenge such as mild overexpressed α-synuclein [
245].
Both NEDD4-1 and NEDD4-2 seem to ubiquitinate, in a PKC-dependent reaction (phosphorylation of NEDD4-2 threonine
197, serine
221, serine
354, and serine
420), the human organic anion transporter 1 (hOAT1) in kidney proximal tubule cells, which is important for the release of anti-HIV drugs, anti-tumour drugs, antibiotics, and anti-inflammatory drugs. Ubiquitination of hOAT1 leads to its reduced activity, internalisation and degradation [
131,
281]. NEDD4-2 has been suggested to also ubiquitinate hOAT3 [
282]. NEDD4-1 seems capable of ubiquitinating the ATP binding cassette transporter B1 (ABCB1), which can export the neurotoxic peptide β-amyloid from endothelial cells in the blood–brain barrier to protect the brain [
31]. In Alzheimer’s patients, ABCB1 protein levels were reduced, while NEDD4-1 protein levels were increased, suggesting NEDD4-1 as a therapeutic target for the treatment of Alzheimer’s disease.
Another interesting NEDD4-1 substrate is the proapoptotic protein RTP801 (regulated in development and DNA damage responses, Redd1; DNA-damage-inducible transcript 4, DDIT4; or dexamethasone-induced gene 2 encoded protein, Dig2), a mTOR suppressor that has previously been shown to cause neuronal death in both cellular and animal models of PD [
85]. RTP801 has been found upregulated in toxin-induced PD animal models such as 6-hydroxydopamine (6-OHDA), MPTP/MPP+ and rotenone, as well as in the substantia nigra dopaminergic neurons of PD patients, while NEDD4-1 seems to be downregulated [
283]. An in vitro study showed that RTP801 can be subjected to lysosomal degradation and is conjugated with K63-linked polyubiquitin chains by NEDD4-1 [
85]. It has been proposed that RTP801 is stress-induced upregulated at the early stage of PD to maintain cellular function, but sustained elevation and mTOR and AKT inhibition might lead to dopaminergic cell death [
284]. However, it has also been suggested that NEDD4-1 acts as a downstream target of the PI3K/PTEN–mTORC1 signalling pathway to promote neurite growth and regeneration [
182].
As ion channel dysfunction become increasingly intertwined with PD pathology, it is worth investigating the capacity of NEDD4 ligases to interact with them, particularly in vivo. Dopaminergic neurons are characterised electrophysioligically by their spontaneous discharge from pacemaker activity to burst-firing [
285,
286,
287,
288,
289,
290]. Tonic action potential firing which contributes to the pacemaker activity of dopaminergic neurons is controlled by ion channels [
291]. Ion channels can play critical roles in the neuronal excitability, cell volume and the regulation of neurotransmitter release.
Voltage-gated sodium channels (Na
vs) reside in the cell membrane are essential for the creation and propagation of action potentials. Studies examining the interaction of NEDD4-2 with Na
vs have been carried out in vivo, in vitro and ex-vivo and demonstrated that Na
vs 1.2, 1.3, 1.5, 1.6, 1.7, 1.8 are substrates of NEDD4-2 mediated through interaction with PPSY, PLSY and PGSP motifs [
122,
123,
124]. In a patch-clamp experiment, Na
v1.2 was shown to be downregulated when NEDD4-2 was expressed in HEK293 cells [
122]. Na
v1.6 is essential for neuronal excitability and a number of motor disorders are associated with mutations in Na
v1.6. Homozygous-null mutations in Na
v1.6 lead to juvenile mortality in mice between P19 and P21 [
292]. Phosphorylated Pro-Gly-Ser
553-Pro motif on Na
v1.6 is a putative binding site for NEDD4 ubiquitin ligases. One study hypothesised that NEDD4-2 contributes to the ubiquitination and subsequent internalisation of Na
v1.6. In cultured cells, NEDD4-2 was indeed shown to interact with Na
v1.6 through a C-terminal Pro-Ser-Tyr
1945 motif, causing a reduction in Na
v1.6 current density. This regulation appears to require both the Pro-Gly-Ser-Pro motif in L1 and the Pro-Ser-Tyr motif in the C terminus of Na
v1.6. When NEDD4-2 binding to the Pro-Ser-Tyr motif was prevented, a stress-mediated increase in Na
v1.6 current density was observed. Phosphorylation of the Pro-Gly-Ser-Pro motif in the L1 of Na
v1.6 seems necessary for stress-induced current modulation. Positive or negative regulation appears to depend on the availability of the PRO-Ser-Tyr motif in the C-terminus to bind NEDD4-2 [
124].
A study examined cognitive impairments in a rat model of PD [
293]. In these 6-OHDA lesioned rats, Na
v1.1 was substantially elevated in reactive hippocampal astrocytes 28 days after lesioning, which reduced after 49 days. No changes were observed in Na
v1.6 levels at 28 days, but was elevated in hippocampal neurons at a later time-point of 49 days post-lesion. The predominantly embryonically expressed Na
v1.3 appeared to be re-expressed in hippocampal CA neurons at 49 days post-lesion. In this study 6-OHDA lesioned rats were treated with the Na
v blocker, phenytoin. These rats exhibited improved spatial learning and memory in the Morris water maze compared to lesioned rats not given the phenytoin [
293].
Navs, in particular Nav1.6, appear to play an important role in neuronal physiology and may play a role in the genesis of congnitive defecits in PD. It would be of interest to examine the in vivo role of Navs in dopaminergic neurons specifically and how this might be altered in the presence or absence of NEDD4 ligases. It would also be of interest to investigate how these channels behave in α-synuclein-mediated models of PD and how this could impact the fate of dopaminergic neurons and their interaction with proximal glia.
In addition to voltage-gated sodium channels, NEDD4 ligases have also been demonstrated to interact with potassium ion channels. In the brain KCNQ2 and KCNQ4 potassium ion channels are mostly limited to the substantia nigra and ventral tegmental area of the midbrain [
294,
295,
296,
297]. In the striatum, dopaminergic nerve termini express KCNQ2 and KCNQ3 [
297]. The muscarine-sensitive K(+) current (M-current) stabilises neuronal resting potential, therefore limiting neuronal excitability. M-current is mediated through heteromeric ion channels comprised of KCNQ3 subunits which associate with either KCNQ2 or KCNQ5 subunits. In a study examining the regulation of KCNQ2/3/5 channels it was revealed that NEDD4-2 but not NEDD4-1 could reduce K(+) currents mediated by KCNQ2/3 and KCNQ3/5 in a
Xenopus oocyte expression system. Through deletion experiments it was shown that the KCNQ3 subunit is required for NEDD4-2 to regulate the heteromeric channels. Co-immunoprecipitation and Glutathione S-transferase fusion pulldown experiments demonstrated that NEDD4-2 and KCNQ2/3 interact directly. NEDD4-2 was also able to ubiquitinate KCNQ2/3 in transfected HEK293T cells [
119]. Other Potassium channels in this family have also been shown to interact with NEDD4 ligases (particularly NEDD4-2) [
116,
120,
121], however these channels have not currently been implicated in PD or dopaminergic system pathology. It appears however that in the nervous system NEDD4-2 is potentially an important M-current activity regulator.
6. Conclusions and Future Directions
The collected data support the notion that further research is required to clarify the unique and common features of NEDD4-1 and NEDD4-2 especially in the midbrain dopaminergic system affected in PD. The large number of possible mechanisms for regulating NEDD4 ubiquitination activity, substrate specificity, and protein interactions make an in silico prediction of possible outcomes extremely difficult. The regulation, function, and substrate specificity of NEDD4-1 and NEDD4-2 need to be studied in vivo in a tissue- and cell-type-specific fashion before strategies can be designed to propose them as therapeutic targets for neurodegenerative diseases such as PD. The possible substrates suggest that NEDD4-1 and/or NEDD4-2 could be beneficial or harmful in the disease context. Currently, it is not clear if NEDD4-1 and/or NEDD4-2 protein levels should be increased or decreased to improve the conditions in dopaminergic neurons under pathophysiological conditions such as PD. Research on NEDD4 proteins in neurodegenerative diseases remains an exciting field in which many surprising findings can still be expected. Unbiased approaches should therefore be applied to remain open to all possible outcomes. In the recent years, the toolkit for studying NEDD4-1 and NEDD4-2 has dramatically improved, with the availability of conditional animal models for NEDD4-1 and NEDD4-2, good antibodies, and specific knockdown possibilities, which will facilitate further investigations.