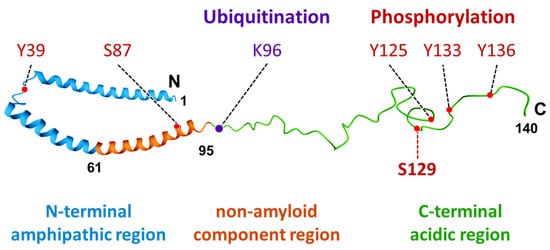
α-Synuclein is a protein with a molecular weight of 14.5 kDa and consists of 140 amino acids encoded by the
SNCA gene. The protein consists of three domains: the N-terminal amphipathic region, the non-amyloid component region, and the C-terminal acidic region (
Figure 1). In the central nervous system, it is particularly abundant in presynaptic terminals and is presumed to be involved in regulating synaptic function and neural plasticity. Synuclein was first identified in cholinergic synaptic vesicles derived from the electric organ of the northeastern Pacific electric ray (
Torpedo californica) [
1]. The name “synuclein” originated from its distribution because its expression was observed not only in synaptic vesicles in the presynaptic terminal but also in the nucleus following immunostaining. Owing to this, α-synuclein was presumed to be a molecule related to the nervous system, especially in synaptic function.
α-Synuclein is particularly highly expressed in the central nervous system in human adults—accounting for approximately 1% of soluble proteins in the brain—and is abundant in presynaptic terminals [
2]. α-Synuclein protein is also abundant in erythrocytes [
3]. Most α-synuclein is distributed in the cytoplasm of cells and its carboxyterminal region, which is flexible in monomeric and fibrillar states and plays an essential role in the interaction with other partners [
4]. Some of the protein is associated with biological membranes, such as synaptic vesicles [
5]. In addition, α-synuclein is observed in the mitochondrial inner membrane, the mitochondria-associated endoplasmic reticulum membranes, the Golgi apparatus, and endosomes [
6,
7,
8,
9]. It also interacts with various proteins, including tau, ATPases, lipid membranes, lymphocyte-activation gene 3, flotillin, and fatty acid-binding protein 3 [
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20], as well as metal ions [
21,
22]. Nuclear α-synuclein presumably functions as a DNA-binding protein and is thought to be involved in regulating various gene expressions [
23,
24,
25].
2. Kinases That Phosphorylate α-Synuclein and Their Association to Cytotoxicity
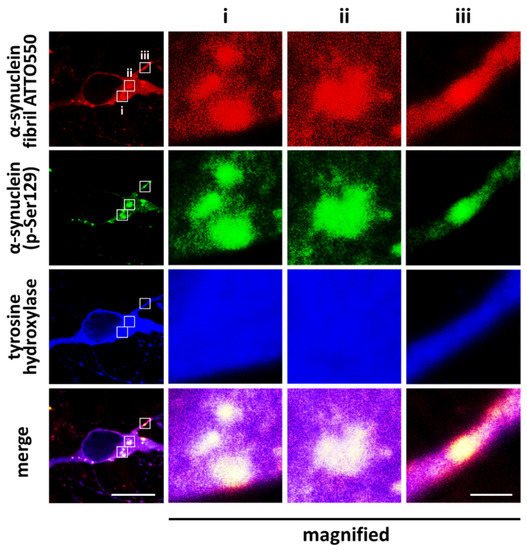
Exogenous α-synuclein that is propagated and taken up into the cell facilitates aggregation with intracellular proteins to form inclusions, which are immunopositive for α-synuclein phosphorylated at Ser129 (
Figure 2). Nonetheless, the question of how α-synuclein obtains these aggregation characteristics and enhances cytotoxicity arises. Earlier investigations reported that α-synuclein within Lewy bodies is subject to post-translational modifications, including phosphorylation and ubiquitination. Notably, among these modifications, the phosphorylation at residue Ser129 and its possible implication on α-synucleinopathy have had significant research focus and are postulated to play a key role in the regulation of α-synuclein aggregation and neurotoxicity [
37,
38,
39]. In particular, a small portion of α-synuclein (~4%) is constitutively phosphorylated at Ser129 in the brains of healthy individuals [
40,
41,
42], whereas >90% of the insoluble α-synuclein is phosphorylated in the brain of patients with synucleinopathies [
41,
43,
44,
45]. The accumulation of phosphorylated α-synuclein at Ser129 was also observed in animal models of Parkinson’s disease [
46,
47,
48,
49,
50]. These observations indicate the importance of α-synuclein phosphorylation at Ser129 in regulating its aggregation and neurotoxicity.
Figure 2. α-Synuclein Ser129 phosphorylation in cultured dopaminergic neurons. Exposure to exogenous α-synuclein fibrils induces the formation of phosphorylated α-synuclein-positive intracellular aggregates. Cells were exposed to ATTO550-labeled α-synuclein fibrils for 48 h. Scale bar = 10 μm, and 1 μm in magnified images of ROI i, ii, and iii. Images were originally obtained for this review.
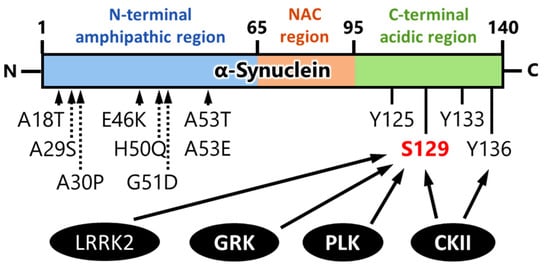
The carboxy-terminal region of α-synuclein is flexible in monomeric and fibrillar forms as well as in membrane-bound states [
4,
18,
51,
52,
53]. Interestingly, the α-synuclein C-terminal tail contains the majority of phosphorylation sites (
Figure 3). Phosphorylation of α-synuclein Ser129 is mediated by several kinases such as G-protein-coupled receptor kinases (GRKs) [
54,
55,
56], casein kinase II [
56,
57,
58], polo-like kinases [
59,
60,
61], and leucine-rich repeat kinase 2 (LRRK2) [
62,
63]. Below, we summarize the α-synuclein phosphorylation kinases and describe the associated pathogenic neurotoxicity.
Figure 3. Summary of representative phosphorylation sites, kinases, and mutation sites of α-synuclein [
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64]. GRK: G-protein-coupled receptor kinase; CKII: casein kinase II; PLK: polo-like kinase; LRRK2: leucine-rich repeat kinase 2; NAC: non-amyloid component.
2.1. Impact of G-Protein-Coupled Receptor Kinases on α-Synuclein Phosphorylation
GRKs are a serine/threonine kinase family that regulate G-protein-coupled receptors. Mammalian GRKs have seven isoforms that can be divided into three subfamilies based on their structural organization and homology: GRK1 (rhodopsin kinase) and GRK7; GRK2 (βARK1) and GRK3 (βARK2); and GRK4, GRK5, and GRK6. Among these isoforms, GRK2 preferentially phosphorylates α-synuclein, while GRK5 prefers α-isoform as a substrate [
54,
55]. Interestingly, GRK-mediated authentic phosphorylation and mimicking of the phosphorylation state (S129E) reduce α-synuclein affinity to binding phospholipids [
54,
65]. Furthermore, phosphomimetic mutation (S129D) inhibits its association with membranes [
66,
67]. These data indicate an inhibitory effect of Ser129 phosphorylation on the α-synuclein binding to the cellular membrane. Following the loss of ability to bind to plasma membrane, phosphorylated α-synuclein preferentially localizes to mitochondria and suppresses cell respiration [
68,
69], which also induces endoplasmic reticulum stress and cell death [
46,
70,
71]. These data indicate that α-synuclein phosphorylation is linked to mitochondrial dysfunction and the production of reactive oxygen species. GRK5 is immunohistochemically co-localized with phosphorylated α-synuclein in Lewy bodies [
55], indicating its pathogenic significance in α-synucleinopathies.
2.2. Impact of Casein Kinase II on α-Synuclein Phosphorylation
Another kinase of α-synuclein phosphorylation, casein kinase II (CK2), is a serine/threonine-selective protein kinase. CK2 is a tetrameric enzyme assembled from two catalytic subunits (CK2α and CK2α′) and a regulatory subunit (CK2β dimer). Casein kinase II is necessary for cell survival and is a crucial suppressor of apoptosis [
72]. Wu et al. reported that CK2α can be S-nitrosylated by nitric oxide (NO). Intriguingly, S-nitrosylation of CK2α enhanced the kinase activity of this catalytic subunit towards the phosphorylation of α-synuclein at Ser129 [
56]. These data indicate that CK2α can enhance the phosphorylation of pSer129 α-synuclein. In addition, Sano et al. found that α-synuclein aggregates are predominantly phosphorylated at Y136 in brains with DLB, which is mediated by the activity of CK2 [
58]. Furthermore, aggregate formation with Ser129 phosphorylation was significantly attenuated by CK2 inhibition, which increased Y136 phosphorylation in cultured neuroblastoma SH-SY5Y cells [
58]. Moreover, CK2β regulatory subunits are detected in the halo region of Lewy bodies in the substantia nigra of patients with Parkinson’s disease [
70]. These data indicate the involvement of CK2-related α-synuclein phosphorylation in the pathogenesis of Lewy-body-associated diseases.
2.3. Impact of Polo-like Kinase on α-Synuclein Phosphorylation
The third phosphorylation candidate, polo-like kinase, is also a regulatory serine/threonine kinase containing an N-terminal kinase catalytic domain and a C-terminal polo-box domain (PBD) that is involved in substrate binding and regulation of kinase activity. Five mammalian PLK family members have been identified, viz. PLK1–5 [
71]. The polo-like kinases PLK2 and PLK3 have phosphorylation activity against α-synuclein that is better than that of PLK1 and PLK4 and are capable of completely phosphorylating α-synuclein [
59,
60]. PLK5 lacks a functional kinase domain due to a premature stop codon in exon 6 and is thus unable to phosphorylate α-synuclein. Interestingly, a significant reduction in α-synuclein phosphorylation has been observed in primary cortical neurons and the cerebral cortex of PLK2 knockout mice [
59]. Bergeron et al. further demonstrated the ability of PLK2 to phosphorylate α-synuclein at Ser129 in the brain of mice in vivo [
61]. These data show that polo-like kinase is critical for α-synuclein phosphorylation in the pathogenesis of α-synucleinopathy. Furthermore, dephosphorylated α-synuclein binding inhibits the activity of tyrosine hydroxylase (TH) [
73], the rate-limiting enzyme in dopamine biosynthesis. Interestingly, phosphorylation of α-synuclein at Ser129 by PLK2 in vitro reduced the ability of α-synuclein to inhibit TH [
74]. Active phosphorylated TH is easily aggregated [
75,
76] and degraded by the ubiquitin-proteasome system [
77,
78], indicating the attenuation of dopaminergic function. These data suggest the relevance of PLK2 in the reduced dopamine levels observed in Parkinson’s disease in an α-synuclein phosphorylation-dependent manner.
3. Pathological Impact of Phosphorylated Synuclein and Its Potential as a Novel Therapeutic Target
Previous investigations involving immunohistochemical and biochemical analyses have accumulated much data on Lewy bodies, showing the deposition of α-synuclein phosphorylated at Ser129 in postmortem brains of patients with Parkinson’s disease and DLB. Thus, it is worth investigating when the accumulation of phosphorylated protein begins in the various stages of Lewy body diseases. In this section, we summarize when phosphorylation at Ser129 occurs during the pathogenesis of Lewy body diseases and propose potential therapeutic targets to modulate α-synuclein phosphorylation.
Previously, Walker et al. reported that a biochemical analysis revealed progressive accumulation of p-Ser129 immunoreactivities as well as a positive correlation between phosphorylation level and the severity of symptoms of Parkinson’s disease and DLB [
101]. Moreover, the accumulation of insoluble phosphorylated forms and the formation of p-Ser129-positive insoluble species became detectable only at the late stages of the diseases, stages IV and V, according to the Unified Staging System [
101,
102]. Zhou et al. also reported a dramatic accumulation of p-Ser129-positive inclusions in various brain regions at the late stages of the disease using postmortem brain samples from patients with Parkinson’s disease [
103]. These findings revealed that pathogenic deposition of insoluble α-synuclein phosphorylated at Ser129 primarily occurs at the advanced stages of synucleinopathies.
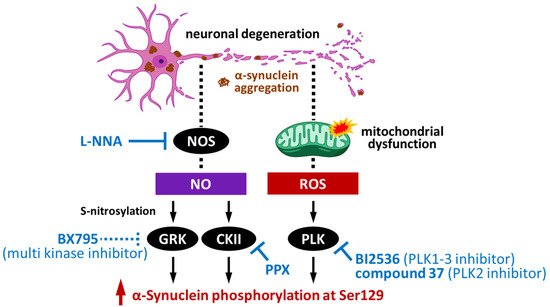
Many kinases are activated in Parkinson’s disease and related disorders and are associated to the diseases’ pathogenesis. Focusing on the kinases that have the ability to phosphorylate α-synuclein (
Figure 3), GRK, CK2, and PLK indeed alter the activity by their modification in Lewy body diseases. As introduced in
Section 4, GRK6 and CK2α can be S-nitrosylated by NO, with S-nitrosylation enhancing their kinase activity towards the phosphorylation of α-synuclein at Ser129 [
56]. Notably, in an A53T α-synuclein transgenic mouse, a familial Parkinson’s disease model, Wu et al. demonstrated that increased GRK6 and CK2α S-nitrosylation was age-dependent and associated with an increased level of p-Ser129 α-synuclein, which suggests GRK6 and CK2α S-nitrosylation as a potential therapeutic target of α-synucleinopathy. In fact, treatment of A53T α-synuclein transgenic mice with Nω-nitro-L-arginine (L-NNA), a NO synthase inhibitor, significantly reduced the S-nitrosylation of GRK6 and CK2α in the brain [
56]. Additionally, Chau et al. reported that pramipexole (PPX) decreases the phosphorylation of CK2 to reduce its activity, thereby inhibiting the phosphorylation of α-synuclein at Ser129 [
104]. Observation of LRRK2 phosphorylation by casein kinase 1α [
105] also raises the possible development of another therapeutic agent.
PLK is also associated with the pathogenesis of Parkinson’s disease. Oxidative stress generated as a consequence of mitochondrial dysfunction can activate PLK2 [
106]. PLK2 knockout reduces presynaptic terminal α-synuclein Ser129 phosphorylation [
107]. In this condition, PLK2 deletion has no effect on the phosphorylation levels in Lewy-body-like inclusions [
107], suggesting that the formation of inclusions is α-synuclein-phosphorylation-independent. Furthermore, PLK2 deletion slows the rate of neuronal death [
107], indicating that inhibition of PLK2 is a potential therapeutic target. Treatment with the PLK inhibitor BI2536 inhibited α-synuclein phosphorylation at Ser129 [
59]. Furthermore, treatment with a designed highly selective inhibitor for PLK2, compound 37, reduced α-synuclein phosphorylation in rat brain [
108,
109]. However, the inhibitor had no effect on α-synuclein aggregation, p-Ser129, or inter-neuronal spreading of the aggregated α-synuclein in the S129A preformed fibril injection model [
110]. These findings suggest that PLK2 could be a potential target for the development of novel therapies, although further optimization is still required at the current stage (
Figure 4).
Figure 4. Schematic pathways of α-synuclein phosphorylation by kinases in the neurodegeneration process and potential pharmacotherapeutic targets [
56,
104,
106,
107,
108,
109,
110]. NO: nitric oxide; NOS: NO synthase; ROS: reactive oxygen species; L-NNA: N(omega)-nitro-L-arginine, a competitive inhibitor of nitric oxide synthase; GRK: G-protein-coupled receptor kinase; CKII: casein kinase II; PLK: polo-like kinase; PPX: pramipexole.