Initial genetic studies based on the use of cytogenetics and FISH helped identify some recurrent chromosomal alterations in T-ALL at the time of diagnosis, but it proved difficult to determine their prognostic impact because of their low incidence in the specific T-ALL cohort analyzed. Genetic knowledge flourished with the application of genomic techniques, first with the analysis of gene expression profiles (GEPs), followed by the application of comparative genomic hybridization arrays (CGHas), and later, the next generation sequencing (NGS) technique. Consequently, we now have a clearer appreciation of the different T-ALL genetic subtypes at the time of diagnosis and are beginning to understand relapse-specific mechanisms.
2. T-ALL Classification by Differentiation Stage
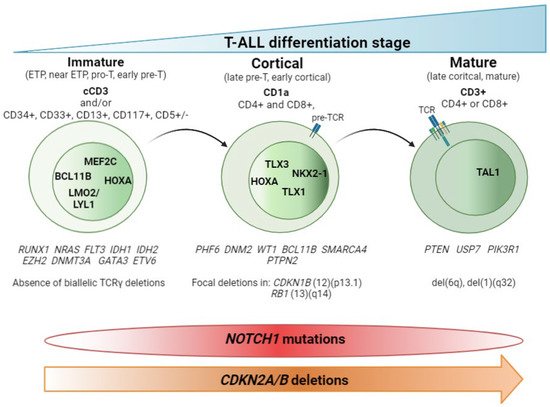
The main contribution of genomic techniques in T-ALL has been to show that the blockade of the differentiation process that occurs in the lymphocyte is the consequence of specific genetic alterations occurring in pre-leukemic stages. From a historical perspective, the first studies to classify T-ALL aroused with the use of the gene expression array (GEa). This revealed that structural abnormalities, mainly rearrangements identified in T-ALL by karyotyping as a rare event [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19], led to much overexpression of (1) basic helix-loop-helix (bHLH) transcription factor genes such as
TAL1,
TAL2, and
LYL1; (2) LIM-only (
LMO) domain genes such as
LMO1 and
LMO2; and (3) homeobox genes such as
TLX1 (
HOX11) and
TLX3 (
HOX11L2). Supervised analysis of the expression data revealed a correlation between the expression of these transcription factors (TFs) and a lymphocyte-specific differentiation arrest time point. Three main groups (clusters) were obtained: (1) immature, (2) HOXA, and (3) TAL [
20,
21]. To describe this in more detail, HOXA samples cluster with the Pre-T1 (CD34 + CD1a+) and Pre-T2/Pre-T3 (CD4+, CD8αβ-, CD3- and TCR αβ-) sub-populations, together with the
TLX1-expressing cases and some cases expressing
TLX3. TAL samples cluster with subpopulations corresponding to thymocytes with a pre-TCR (Beta selection). Of note, samples from the immature group included some TLX3-expressing leukemic samples and clustered with the most immature early T-cell precursor and pro-T subpopulations [
21]. Later on, the addition of copy number data, generated by CGHa, together with the use of NGS, yielded a clearer view of the genetic determinants that define these groups. Thus, according to the differentiation arrest time point of the blast cell we can differentiate the immature subtype, characterized by the absence of CD8 and CD1a immunomarkers, high levels of expression of
LYL1 [
20] and
MEF2C [
22] TFs, and the absence of bi-allelic deletions in TCRγ [
23]. Within the immature T-ALL leukemias, the early T-cell precursor ALL (ETP-ALL)—defined by the absence of CD1a and CD8 immunomarkers and the presence of stem cells or myeloid markers such as CD117, CD34, HLA-DR, CD13, CD33, CD11b, and CD65 [
24], together with negative or dim CD5 expression, defined as expression in <75% of the blasts; rarely presents
CDKN2A/B deletions and
NOTCH1/FBXW7 mutations [
25,
26,
27]. In addition, mutations affecting epigenetic regulators and transcription factors governing hematopoietic and T-cell development are also frequently observed in this immature subtype [
25,
28,
29,
30,
31]. We will discuss this subgroup in detail later. The cortical subtype is characterized by the expression of CD1a, and often both CD4 and CD8 immunomarkers are also found. At the genetic level, the group is characterized by aberrant expression of
TLX1,
TLX3, and
HOXA genes (
HOXA5,
HOXA9,
HOXA10) [
20,
21] and the overexpression of the
NKX2-1 rearranged gene [
22]. Other specific alterations in this subset of T-ALL have been found in
PHF6,
DNM2,
BCL11B,
CDKN1B, and
RB1 [
32]. The mature subtype is characterized by blasts expressing CD4 or CD8 and surface CD3 immunomarkers [
20] and the presence of Tαβ cell-receptor rearrangements [
21]. The genetic hallmark of the group is the activation of the
TAL oncogene [
20], together with the presence of del(6)(q) [
33] and mutations in
PIK3R and
PTEN [
32].
Figure 1 summarizes these findings.
Figure 1. T-ALL classification by stage of differentiation arrest. Schematic representation of the three main T-ALL subtypes according to the blockade of the differentiation process. Hallmark immunomarkers of each subtype are highlighted in bold at the top. Other accompanying immunomarkers, for precise definition of the subtypes, are showed below. TCR maturation is represented in the blasts. Presence of TCR on the blast surface is also a hallmark in the mature subtype. Active transcription factors in each subtype of T-ALL are represented in the nucleus according to the maturation transition. The genes most frequently mutated in each subtype, followed by copy number alterations, are shown beneath. The distributions of NOTCH1 mutations and CDKN2A/B deletions are indicated at the bottom.
Collectively, the data generated during twenty years of genomics research into T-ALL highlight the importance of using high-resolution genetic techniques. These not only make it possible to detect the cryptic aberrations present in T-ALL but also to define primary genetic events that determine the acquisition of the secondary genetic events necessary for transforming T-cell progenitors. This thereby explains the particular oncogenetic processes taking place in each T-ALL. We are moving towards an immuno-genetic T-ALL classification. One of the knowledge gaps here relates to T-ALL leukemias with unidentified primary events, although the driving TF is aberrantly expressed. In those cases, it is reasonable to argue that alterations in regulatory regions and/or in enhancers of the TF can alter its expression.
2.1. Non-Coding Mutations
Analysis of non-coding data generated by whole genome sequencing (WGS) or direct target sequencing is an emerging area of investigation, but recently published data have shown that small (or not) insertions and deletions generating novel regulatory sequences can explain the overexpression seen in TFs, such as TAL1 and LMO1, that have no detectable primary rearrangements. Here we provide a summary of the non-coding alterations identified in TAL1, LMO1/2, and other important T-ALL oncogenes such as MYC and PTEN. Non-coding variants discovered in T-ALL are also summarized in Table 1.
Table 1. Non-coding mutations identified so far in T-ALL.
| Gene |
Affected
Region |
Variant |
Alteration |
Functional
Impact |
Frequency |
Reference |
| MYC |
1427 kb
downstream of MYC |
Focal
duplications |
Creation of binding site for NOTCH1 |
MYC
expression |
8/160 (5%)
Adult and
pediatric |
[34] |
| TAL1 |
8 kb upstream of the transcription start site of TAL1 |
Heterozygous indel (2–18 bp) |
Creation of binding motifs for the MYB TF |
TAL1
overexpression |
8/146 (5.5%)
pediatric |
[35,36] |
| LMO1 |
4 kb upstream of the transcriptional start site of LMO1 |
SNV: C → T |
Creation of binding motifs for the MYB TF |
LMO1
overexpression |
4/187 (2.14%) pediatric |
[36,37] |
| LMO2 |
Non-coding region of the exon 2 of LMO2 |
Heterozygous
indel |
Creation of binding motifs for the MYB TF |
Activating LMO2 function |
6/160 (3.75%)
pediatric
9/163 (5.52%)
adult |
[38] |
| PTEN |
550 kb downstream of transcription start site of PTEN |
Focal deletions |
Deletion of PTEN enhancer region |
Reduced levels of PTEN |
5/398 (1.25%) |
[39] |
Abbreviations: MYC: MYC proto-oncogene, bHLH transcription factor; TAL1: T cell acute lymphocytic leukaemia 1; bp: base pair; LMO1: LIM domain only 1; SNV: single nucleotide variant; TF: transcription factor; LMO2: LIM domain only 1; PTEN: phosphatase and tensin homolog; PE: PTEN enhancer.
The first non-coding alterations found to affect T-ALL corresponded those affecting
MYC TF activation by
NOTCH1 [
34]. Recurrent focal duplications at chromosome 8q24 were identified in a CGH screening in 8/160 (5%) of the T-ALL cases analyzed. The amplification was located +1427 kb downstream of
MYC. The alteration was named N-Me for NOTCH
MYC enhancer. The region was shown to interact with the
MYC proximal promoter and induced orientation-independent
MYC expression in reporter assays. Analysis of N-Me knockout mice demonstrated a selective role of this regulatory element in the development T-ALL in a NOTCH1-induced T-ALL model [
34].
Concomitant to this finding, a non-coding alteration located in a regulatory region of
TAL1 was also identified. A 12-bp indel that introduced two consecutive de novo binding motifs for the MYB TF, creating a super-enhancer, was identified using chromatin immunoprecipitation experiments and sequencing (CHIP-seq) in the Jurkat cell line [
35]. The screening of 146 unselected pediatric primary T-ALL samples collected at diagnosis revealed that eight cases (5.5%) contained the 2–18-bp heterozygous indel, confirming the in vitro results. Sequencing DNA from remission bone marrow samples in two available cases showed wild-type sequences at this site, indicating that the mutations were somatically acquired in the blasts. The authors suggested that the initial mechanistic event in the aberrant super-enhancer formation was the recruitment of CBP by MYB, followed by abundant H3K27 acetylation, which facilitated the binding of a core complex composed of RUNX1, GATA-3, and
TAL1 itself [
35]. These results were validated in a study that set out to identify non-coding mutations in 31 pediatric T-ALL cases from the available WGS data. Non-coding
TAL1 mutations were significantly associated with
TAL1 expression, implying a cis-regulating effect. Consistent with previous results, a similar, MYB binding-dependent
TAL1 promoter activation mechanism was described [
36].
Subsequent studies used the same experimental procedure; (1) identification of a core sequence by CHIP-seq in T-ALL cell lines; (2) identification of epigenetic marks supporting transcriptional activity in the region and the transcriptional complex; and (3) validation of the alteration in a large cohort of pediatric T-ALL cases revealed a C-to-T single nucleotide transition occurring as a somatic mutation in the non-coding sequence 4 kb upstream of the transcriptional start site of the
LMO1 TF. This single nucleotide alteration gives rise to an APOBEC-like cytidine deaminase mutational signature and generates a new binding site for the MYB transcription factor, leading to the formation of an aberrant transcriptional enhancer complex that drives high levels of expression of the
LMO1 oncogene, similar to the
TAL1 super-enhancer. Sequencing 187 pediatric primary T-ALL samples collected at diagnosis identified four patients (2.14%) with the same heterozygous mutation [
37]. As with the
TAL1 indel, the
LMO1 aberrant MYB-dependent super-enhancer was confirmed by the WGS study [
36]. However, Shaoyan and colleagues also identified an intrachromosomal inversion event that juxtaposed the active promoter of the
MED17 gene with the coding sequence of
LMO1, leading to the expression of
LMO1. This highlights how other structural abnormalities may help explain the abnormal expression of this TF [
36]. In the case of
LMO2, a heterozygous 20-bp duplication in PF-382 cells and a heterozygous 1-bp deletion in DU.528 cells were identified in the same way, both being located closer to a region recently described as an intermediate promoter. These alterations were not limited to T-ALL cell lines, since heterozygous mutations in the
LMO2 intron 1 were detected in diagnostic samples from 6/160 of the pediatric and 9/163 of the adult T-ALL cases sequenced [
38]. The confirmatory WGS study, in this case, confirmed the non-coding mutations in regulatory regions of
LMO2. However, in this case, they were not associated with
LMO2 gene expression [
36] (
Table 1).
An intronic sequence of 550-kb situated in the neighborhood of the
RNLS gene and downstream of the
PTEN gene has very recently been identified and found to interact strongly with the
PTEN promoter. The presence of enhancer marks in this region, including high levels of H3K27ac and H3K4me1, together with binding of CTCF, BRD4, and ZNF143, defined this region as a
PTEN enhancer (PE). Screening for genetic lesions in this region in human primary samples identified five cases (5/398, 1.25%) with focal deletions encompassing PE. The deletion was homozygous in two of these samples, and additional simultaneous deletions targeting the coding region of
PTEN were observed in four of the five cases. Analyses of 1415 BCP-ALL samples failed to identify the same deletion, showing that these alterations are restricted to the T-ALL subtype [
39] (
Table 1).
Identification of non-coding variants is a burgeoning area of research in which much information is yet to be gathered. The contribution of non-coding sequences to oncogenetics remains largely unknown. In addition to alterations in promoters, regulatory regions and enhancers, as well as other non-coding regions such as intergenic and splicing site sequences, will help refine the immuno-genetic T-ALL classification.
2.2. T-ALL Related Immature Subtypes
Application of WGS and whole transcriptome sequencing has also served to better characterize rare subtypes such as the immature T-ALL leukemias and to provide insight into the cell of origin of these subtypes. This group includes T/Myeloid mixed phenotype acute leukemias (T/M MPALs) and ETP-ALL, which are characterized by different combinations of myeloid and T-lymphoid antigen expression [
40]. Other immunophenotypically identified immature T-ALL subtypes include the pro-T [
40] and the near-ETP [
32,
41] forms, but we do not know their genetic basis or clinical implications. Thus, childhood ETP-ALL presents cytokine-activating somatic mutations and mutations in genes involved in the RAS signaling pathway (e.g.,
NRAS,
KRAS,
FLT3,
IL7R,
JAK3,
JAK1,
SH2B3, and
BRAF), genetic alterations that inactivate genes involved in hematopoietic development (e.g.,
GATA3,
ETV6,
RUNX1,
IKZF1, and
EP300), and mutations in histone modifier genes (e.g.,
EZH2,
EED,
SUZ12,
SETD2, and
EP300). It is of note that the mutational spectrum identified in ETP leukemia was similar to that of acute myeloid leukemia (AML) with poor prognosis, in which affected pluripotent genes lend this subtype a myeloid-like profile [
29]. In the case of adult ETP-ALL, exclusively genetic alterations have been detected in the
DNMT3A gene (frequency range from 12 to 16%) [
30,
31,
42,
43], in addition to the aforementioned mutations. Specifically,
DNMT3A mutations are associated with patients aged >60 years with ETP-ALL features [
43].
FAT1 (25%, 17/68) and
FAT3 (20%, 14/68) cadherins are other mutations exclusively found in adult ETP-ALL [
30] (
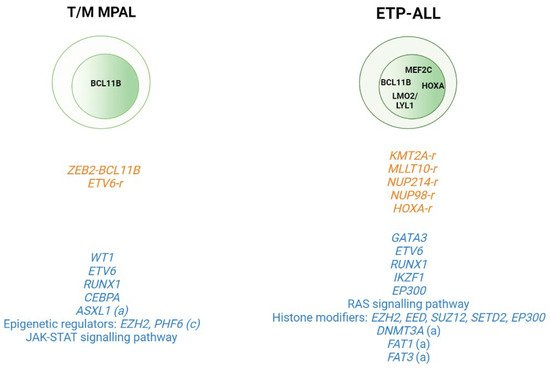
Figure 2). In addition to point mutations, structural abnormalities such as rearrangements affecting
KMT2A,
MLLT10,
NUP214, or
NUP98, which trans activate
HOXA genes, are often detected in ETP-ALL cases [
44,
45]. Overexpression of the
BCL11B gene due to different structural abnormalities including translocations (i.e., t(2;14)(q22.3;q32), t(6;14)(q25.3;q32), hijacking super-enhancers, and other fusion genes has been very recently described as present in one third of ETP-ALL and T/myeloid mixed phenotype acute leukemia (T/M MPAL) cases with a very distinct expression profile [
46,
47].
Figure 2. Genomic alterations in T/Myeloid mixed phenotype acute leukemias (T/M MPALs) and ETP-ALL. Active transcription factors in each subtype are represented in the nucleus according to the maturation transition. The rearrangements and fusion genes are written in yellow and most frequently mutations in blue color. (a) adult; (c) children.
In the case of T/M MPAL leukemias, a study cohort including 49 pediatric cases of this mixed phenotype showed a high number of copy number alterations (CNAs) (average of 4.5 (0–35)) in this subtype compared to KMT2Ar MPAL leukemias. Alterations in genes encoding transcriptional regulators were also detected in the T/M MPAL cases (i.e., WT1,
ETV6,
RUNX1, and
CEBPA) [
48]. Alterations in JAK–STAT signaling were also common in these leukemias together with mutations in genes encoding epigenetic regulators (69% of cases), including inactivating mutations in
EZH2 (16%) and
PHF6 (16%). Analysis of the transcriptome sequencing identified chimeric in-frame fusions in 15/40 cases, including
ZEB2–
BCL11B (
n = 3) and several fusions involving the
ETV6 gene [
48].
Comparison of the genetic profiles of ETP-ALL and T/M MPAL with non-immature T-ALL leukemias in pediatric cases has shown that the core TF driving T-ALL (
TAL1,
TAL2,
TLX1,
TLX3,
LMO1,
LMO2,
NKX2- 1,
HOXA10, and
LYL1) is less frequently altered in T/M MPAL and ETP-ALL. Other alterations that are common in T-ALL, such as
MYB amplification,
LEF1 deletion, and
CDKN2A/B deletions, are also rare in both types of immature leukemia. By contrast,
WT1 alterations are common in T/M MPAL and ETP-ALL, but not in non-immature T-ALL [
48]. A similar analysis conducted in adult MPAL leukemias observed that, while myeloid-T/M MPAL and T-ALL shared a number of mutations in common, there were also differences.
PHF6 and
JAK3 mutations, each detected in 21.4% of the adult T-ALL cases analyzed, were not detected in myeloid-T/M MPAL. In contrast,
ASXL1 (11.1%) and
FLT3 (11.1%) mutations were detected in T/M MPAL but not in T-ALL [
49]. Collectively, these observations imply that different primary events can drive specific T-ALL subtypes. A summary of these data can be seen in
Figure 2.
3. (Epi)genetic Modification
The systematic screening of T-ALL genomes has revealed T-ALL as one of the tumors with the highest frequency of mutations in genes that encode proteins involved in epigenetic regulation [
50]. Hence, the field of epigenetics, particularly DNA methylation, is currently being extensively explored in the search for specific methylation patterns that help to explain the oncogenic evolution of pre-leukemic T-cells; to identify specific de-regulated genes to use as a prognosis marker; and to delineate new therapy strategies using DNA methylation inhibitors (iDNMTs) such as 5-azacitidine (vidaza, AZA) and 5-aza-20 -deoxycytidine (decitabine, DAC) [
51].
Initial epigenetic studies were focused on determining the methylation status of the promoter of specific genes playing a role in the T-ALL oncogenic process such as
CDKN2A/B. It was observed in T-ALL patients that the percentage of promoter methylation in the
CDKN2B and
CDKN2A genes ranged between 46% and 68% and between 0% and 12%, respectively, in pediatric cohorts ([
52,
53,
54]. In the case of adult T-ALL cohorts, the percentage of
CDKN2B gene promoter methylation varied from 16% to 49% and was 1% for the
CDKN2A promoter [
55,
56,
57,
58,
59]. In T-ALL, the
CDKN2B methylation status was associated with an immature immunophenotype [
58] and with ETP-ALL features [
59]. Further investigation of the methylation status in cancer cells using wide genomic approaches (i.e., methylation arrays) have observed that generally malignant cells display a DNA hypermethylation pattern at specific CpG islands; globally, this observation is called a CpG island methylator phenotype (CIMP). CIMP+ in T-ALLs has been associated with a better EF and OS as compared to CIMP− leukemias [
60]. Notably, these findings have been confirmed in both pediatric [
61] and adult [
62] T-ALL cohorts, reinforcing the idea that aberrant DNA methylation might act as a clinically relevant biomarker in human T-ALL. Comparison of the global methylation profile of T-ALL samples with that of normal thymocytes observed that the methylation profile of CIMP− cases was close to normal CD3+ and CD34+ thymocytes [
60,
61]. That was interpreted as an indication of a shorter proliferation history of the CIMP− blasts as compared to CIMP+ cases [
63]. Together, these findings indicate that CIMP− cases are characterized by a hypomethylation pattern that results in a young mitotic age and shorter proliferative history of leukemic cells; at the same time, however, it might be considered as a marker of higher aggressiveness of leukemic cells. In CIMP+ cases, the disease latency is longer, which is reflected by higher methylation acquired during the aging of pre-leukemic cells, [
61,
63,
64]. Altogether, these results indicated that aberrant methylation is likely not a driving force of T-ALL onset and progression but is rather related to the proliferative history of the cells. These concepts have been nicely reviewed by Natalia Mackowska et al. [
65] in a recent publication.