Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The main cause of death in patients with type 2 diabetes mellitus (DM) is cardiovascular complications resulting from the progression of atherosclerosis. The pathophysiology of the association between diabetes and its vascular complications is complex and multifactorial and closely related to the toxic effects of hyperglycemia that causes increased generation of reactive oxygen species and promotes the secretion of pro-inflammatory cytokines. Subsequent oxidative stress and inflammation are major factors of the progression of type 2 DM and its vascular complications
- diabetes
- inflammation
- cytokines
- mitochondria
1. Inflammation in Insulin Resistance and Diabetes Mellitus
Inflammation is seen as a critical factor in metabolic dysregulation. Suppression of inflammatory reactions has been considered a metabolically protective process that reduces the development of insulin resistance and type 2 diabetes mellitus (DM) . Over a century ago, high doses of sodium salicylate were found to reduce glucosuria in people diagnosed with diabetes [1][2]. Insulin resistance of the liver, adipose tissue, and skeletal muscle stimulates the secretion of insulin from the pancreas, which maintains a normal level of glycemia (pre-diabetic stage) [3][4][5]. The effective relationship between insulin-secreting cells and insulin target tissues (pancreas, adipose tissue, liver, and skeletal muscle) maintains metabolic homeostasis in response to physiological fluctuations in glycemia or lipemia, in response to food intake or starvation. Insulin resistance represents a partial disruption of communication between these tissues, in which target tissues of insulin become resistant to insulin signaling despite initial compensation by the pancreas. Type 2 DM is the stage of complete or near complete disorder of this relationship, when insulin production no longer fits the organism’s need for glycemic regulation. Each of these target tissues has its own specialized macrophages to maintain important physiological functions, to keep tissue integrity, and, more importantly, the number of macrophages in the tissue undergoes adaptation at each stage of type 2 DM development [6][7]. Tissue macrophages are extremely potent mediators of insulin signaling, sensitivity, and resistance. Macrophages quickly respond to environmental signals and adapt their functions. To date, an important role of macrophage polarization in the development of metabolic diseases has been established [8].
An important role in insulin resistance of cells is played by the endoplasmic reticulum (ER). When its homeostasis is disturbed, “ER stress” occurs, which is a key sign of metabolic disorders [9][10]. In obesity, which often accompanies type 2 DM, persistent metabolic pressure leads to disruption of essential ER functions and consequently to impaired cellular health, inflammation, and ultimately metabolic collapse [10]. In addition, hyperglycemia, an independent risk factor for type 2 diabetes, leads to a significant induction of ROS, which causes increased activation of inflammatory pathways [11]. In the occurrence of type 2 diabetes, as well as associated cardiovascular complications, such as atherosclerosis, macrophages/monocytes play a key role as the main regulators of inflammation. Numerous studies show the presence of many levels of regulation of these processes, from cell surface receptors to nuclear receptors, transcription factors, and their coregulators [7][12]. In particular, the mechanisms of impaired insulin action are associated with serine/threonine phosphorylation, which mediates insulin receptor (IR) signaling [13]. In addition, inhibitory phosphorylation can be initiated by pro-inflammatory cytokines (TNFα, IL-1β, and IL-6) secreted by macrophages. These cytokines in turn activate serine kinases such as IκB kinase β (IKKβ), c-Jun N-terminal kinase (JNK), ribosomal protein S6 kinase (S6K), and mammalian target of rapamycin 32 (mTOR32) in adipocytes, which mediate inhibitory phosphorylation of insulin receptor substrate 1 (IRS1), causing insulin resistance [14]. The same kinases play an important role in initiating the immune response through the toll-like receptor (TLR), which, upon activation, secretes the production of various cytokines [15][16]. A brief scheme demonstrating the main inflammatory mechanisms in the development of insulin resistance is shown in Figure 1.
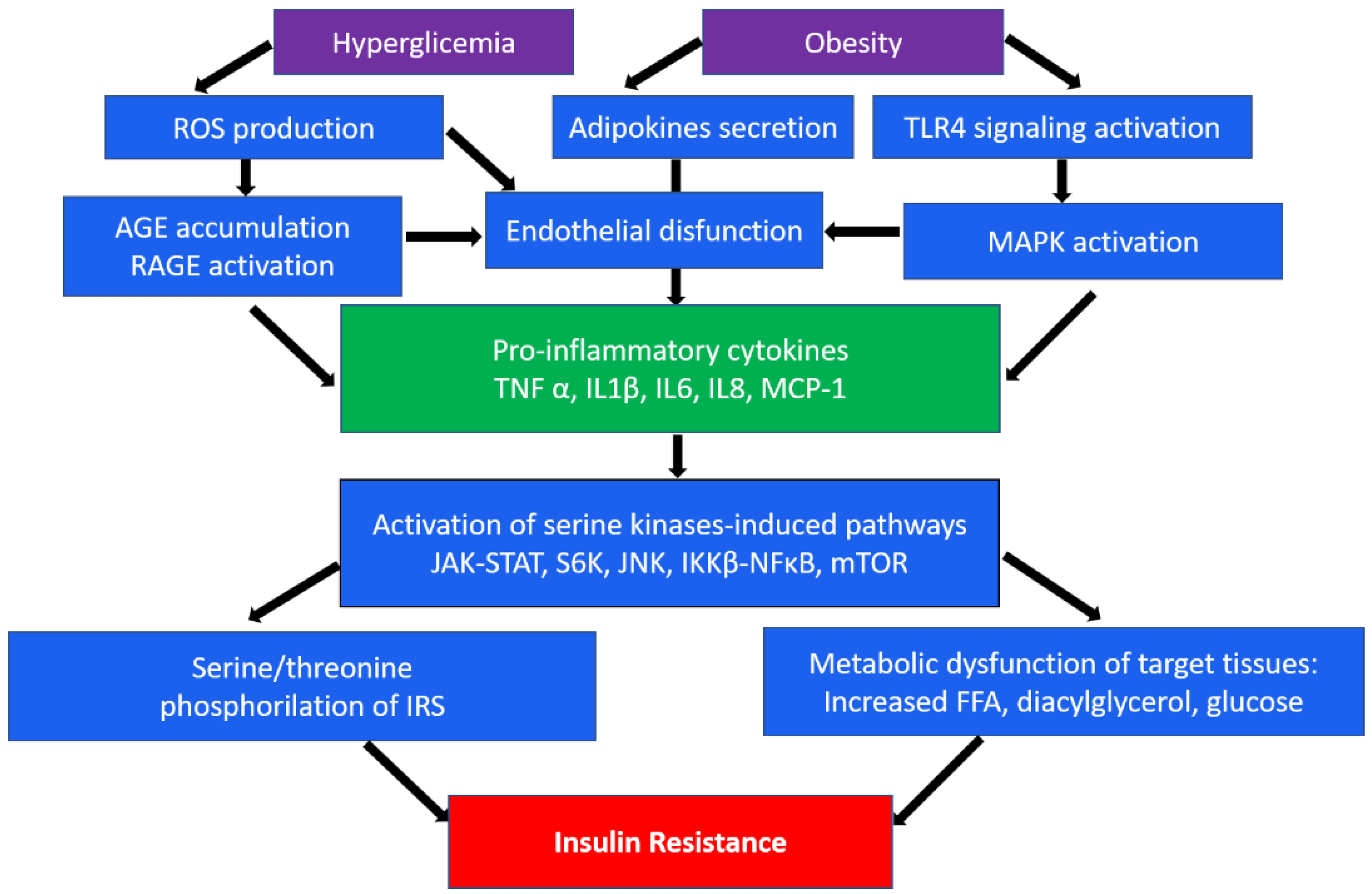
Figure 1. A brief scheme of the main inflammatory mechanisms in the development of insulin resistance. ROS, reactive oxygen species; TLR4, toll-like receptor 4; AGE, advanced glycation end-products; RAGE, AGE receptor; MAPK, mitogen-activated protein (MAP) kinase; TNFα, tumor necrosis factor α; IL, interleukins; MCP-1, monocyte chemoattractant protein-1; JAK-STAT, Janus kinase signal transducers as well as activators of transcriptional signaling pathway; S6k, ribosomal protein S6 kinase; JNK, c-Jun N-terminal kinase signaling pathway; NF-κB, nuclear factor-κB pathway; mTOR, mammalian target of rapamycin; IRS1, insulin receptor substrate 1; FFA, free fatty acids.
It is known that adipose tissue consists mainly of adipocytes, as well as preadipocytes, lymphocytes, macrophages, fibroblasts, and vascular cells. The content of macrophages is greater in visceral than in subcutaneous adipose tissue, which indicates that the accumulation of visceral fat contributes to insulin resistance and can lead to metabolic diseases. Obesity leads to the change in adipose tissue cellular composition as well as to the activation of immune cells [17]. Adipocyte hypertrophy, hypoxia, and increased cell death due to increased accumulation of lipids, in particular triglycerides, contribute to secretion of pro-inflammatory molecules such as TNF-α, IL-6, IL-8, and MCP-1, adipokines and so on, by adipocytes and immune cells, in particular macrophages, resulting in increased infiltration of circulating monocytes and immune cells into adipose tissue [18][19]. Recruited monocytes differentiate into a pro-inflammatory M1 macrophage phenotype, resulting in an imbalance between M1 and M2 macrophages and reduced anti-inflammatory signals from M2 macrophages. This contributes to greater secretion of pro-inflammatory cytokines and adipokines, and consequently, dysfunction of adipose tissue and decrease in glucose tolerance [20]. Most pro-inflammatory stimuli simultaneously activate the JNK and IKKβ TLR pathways. Activation of JNK and IKKβ/NF-κB can occur due to the influence of pro-inflammatory cytokines such as TNF-α and IL-1β through receptor-mediated mechanisms, as well as non-receptor mechanisms through activation of receptors such as TLR and glycation end products receptor (RAGE) recognition patterns, defined as surface proteins that recognize foreign substances. JNK promotes insulin resistance through phosphorylation of serine residues in IRS-1, and IKKβ induces insulin resistance through transcriptional activation of nuclear factor-κB (NF-κB). Activation of the JNK and NF-κB pathways will lead to the production of pro-inflammatory cytokines and mediators, which further activates these pathways via feed-forward mechanisms [21]. Obesity accompanied by dysfunction of adipose tissue plays a decisive role in the pathogenesis of not only insulin resistance but also liver pathologies such as non-alcoholic steatohepatitis (NASH) and non-alcoholic fatty liver disease (NAFLD) [22]. The relationship between NAFLD and type 2 DM is very complex and bidirectional. On the one hand, NAFLD is an independent risk factor for the development of diabetes. On the other hand, diabetes can contribute to the progression of NASH, NAFLD, liver cirrhosis and, in some cases, hepatocellular carcinoma. In addition, NAFLD is a risk factor for cardiovascular disease, especially in combination with type 2 diabetes [23][24]. The pathogenesis of NAFLD involves the “multiple parallel strikes” hypothesis, which involves insulin resistance, liver triglyceride accumulation, oxidative stress, and processes in adipose tissue that promote a cascade of inflammation and cytokine and adipokine production that leads to liver damage [25].
The major inflammatory pathways playing a key role in diabetes development were demonstrated in mice models.
2. Macrophages in Type 2 Diabetes
Currently, it was showed that extracellular, metabolic, and molecular signals associated with macrophage polarization is important in metabolic inflammation and insulin resistance. In obesity, macrophages make up 50% of all adipose tissue cells [26]. Adipose tissue macrophages (ATMs) not only increase in number, but also change their localization, being located around dead adipocytes and forming crown-like structures (CLSs), which exhibit pronounced pro-inflammatory properties [27]. Activation of inflammatory pathways in adipocytes and macrophages is carried out through toll-like receptors, in particular, TLR4. It has been shown that free fatty acids (FFA), which are elevated in obesity, may promote TLR4 signaling, which in turn contributes to obesity-related insulin resistance [28]. In addition, low-density lipoprotein receptors (LDLR) have been shown to be involved in the development of insulin resistance. Increased expression of LDLR in adipocytes of adipose tissue contributes to pro-inflammatory activation and insulin resistance in obesity [29].
Macrophage infiltration into adipose tissue leads to the secretion of pro-inflammatory cytokines such as TNFα, IL-1β, and IL-6, which activate serine kinases in adipocytes, including IKKβ, N-terminal c-Jun kinase (JNK), S6K, and mTOR32. All these kinases trigger inhibitory phosphorylation of IRS1 [14]. Janus kinase (JAK) signal transducers as well as activators of transcriptional (STAT) signaling pathways play an important role in maintaining homeostatic processes. Activation of JAK results in phosphorylation of tyrosine residues in the STAT protein. The JAK-STAT signaling pathway transcriptionally regulates the cytokine signaling suppressor (SOCS), which inhibits JAK and STAT activation and phosphorylation [30].
The general consensus about obesity and type 2 DM is that there is a disbalance in the ratio of M1/M2 macrophages, which leads to an increase in the number of pro-inflammatory M1 macrophages compared to anti-inflammatory M2 macrophages, leading to chronic inflammation and the spread of metabolic dysfunction [6]. Insulin acts on cells through the insulin receptor (IR), located on the surface of insulin-sensitive cells. As a result of IR stimulation, its autophosphorylation occurs, as well as subsequent tyrosine phosphorylation of members of the IRS family, thus initiating signaling events in the cell [31][32]. Dysregulation of insulin signaling, resulting from various factors, is the main mechanism leading ultimately to insulin resistance. In particular, IRS-1 serine phosphorylation leads to increased levels of free fatty acids, diacylglycerol, fatty acyl-CoA, ceramides, and glucose, leading to obesity-associated insulin resistance. Some cytokines, in particular, TNF-α secreted by adipose tissue cells, stimulate the phosphorylation of IRS-1 serine and threonine residues, which reduces IRS-1 tyrosine phosphorylation in response to insulin, as well as the ability of IRS-1 to bind to the insulin receptor, which, in in turn, suppresses signaling [33].
TNF-α and IL-6 increase the expression of SOCS proteins through attenuating insulin signaling by binding to insulin receptors and reducing their ability to phosphorylate IRS proteins. On the other hand, SOCS proteins can directly bind to IRS proteins, resulting in their degradation. In addition, these cytokines can inhibit the expression of IRS-1 at the transcriptional level. Thus, suppression of IRS-1 mRNA expression seems to be the main mechanism involved in altering IRS-1 tyrosine phosphorylation in adipocytes of patients with type 2 DM. IL-1β exerts its pro-inflammatory action by binding to the type I IL-1 receptor and activating the IKK/NF-κB pathway and the three types of mitogen-activated protein (MAP) kinases—extracellular signal-regulated kinase (ERK), JNK, and p38MAPK—which also determines its involvement in insulin resistance [34]. Activation of MAP kinase signaling pathways promotes endothelin-1 (ET-1) secretion, activation of cation pumps, and increased expression of vascular cell adhesion molecule 1 (VCAM-1) and E-selectin. ET-1, in turn, can enhance serine phosphorylation of IRS-1, causing a decrease in PI-3 kinase activity in vascular smooth muscle cells, as well as disrupt insulin-stimulated glucose transporter type 4 (GLUT-4) translocation in adipocytes [35]. It has been shown that IL-1β levels are elevated in non-diabetic offspring of diabetics and correlate with metabolic syndrome, as well as increased expression of both IL-1β and its receptor in visceral adipose tissue in obese individuals [34].
3. The Role of Mitochondria in the Development and Progression of Type 2 Diabetes and Its Vascular Complications
Mitochondria play a key role in metabolic processes in all cells of the body. In endothelial cells, they have a direct effect on the formation of endothelial dysfunction and, therefore, vascular diseases, such as atherosclerosis and diabetic vascular dysfunction, accompanying type 2 diabetes [36][37][38]. The main function of mitochondria is to produce cellular energy using the cyclooxygenase (COX) and associated inner membrane electron transport chain, which produces ATP. In addition, mitochondria are involved in the formation of ROS, calcium and iron homeostasis, steroid biosynthesis, immune cell activation, apoptosis, and inflammation [39][40]. When antioxidant defense mechanisms are disrupted, excessive accumulation of ROS occurs, which not only directly damages cells by oxidizing DNA, proteins, and lipids, but also indirectly damages cells by activating stress-sensitive intracellular signaling pathways, such as NF-κB, p38 MAPK, and JNK/SAPK. Activation of these pathways leads to the development of inflammation and, ultimately, diseases associated with oxidative stress [41][42].
It is known that mitochondria, both in physiological conditions and in pathology, are heterogeneous and have different functional properties, and have differences in morphology, membrane potential, and mitochondrial calcium levels. A pool of mitochondria in an individual cell can represent mitochondria at different stages of development, as well as the consequences of their response to the cellular environment. Mitochondrial heterogeneity is a two-way process in which a lower degree of heterogeneity can be beneficial and can provide the cell with protection and adaptation to biological stress, and a higher degree of heterogeneity can lead to irreversible disease because of the accumulation of dysfunctional or inadequate mitochondria. Studies have described that within the same cell, mitochondria exhibit broad heterogeneity in mitochondrial membrane potential (MMP), which can be generated by the BCL2-associated agonist of cell death (BAD) protein, a member of the proapoptotic BCL-2 family. Glucose-stimulated mitochondrial hyperpolarization has been shown to increase insulin secretion. In addition, mitochondrial heterogeneity provides metabolic flexibility [43].
Under physiological conditions, mitochondria are not static organelles; as a result of continuous cycles of fusion and division, they change their shape and location depending on physiological stimuli. The regulation of mitochondrial dynamics is a complex process that is controlled by several dynamin-related guanosine triphosphate hydrolases (GTPases) that maintain the balance between mitochondrial fusion and fission. Mitofusins MFN1 and MFN2 are responsible for outer mitochondrial membrane (OMM) fusion, while mitochondrial inner membrane (IMM) fusion is regulated by optic atrophy protein 1 (OPA1). Fission proteins include dynamin-related protein 1 (DRP1) and fission protein 1 (FIS1). Mitochondrial fission is essential for the removal of defective mitochondria by mitophagy, which is mediated by (PTEN)-induced putative kinase 1 (PINK1), PARKIN ubiquitin ligase, ubiquitin, and sequestosome-1 (p62/SQSTM1) [44]. It was shown that in patients with type 2 diabetes and obesity, MFN2 expression was reduced, which may be associated with decreased mitochondrial function [45]. Any change in this balance can lead to oxidative stress and mitochondrial dysfunction, including Ca2+ overload, decreased ATP synthesis, and loss of MMP. In turn, this is an important reason for the phenotypic transformation of vascular smooth muscle cells (VSMCs), which can release apoptotic bodies that induce calcification, leading to vessel remodeling and vascular wall stiffness. Ultimately, this can lead to metabolic disorders that underlie the development of type 2 diabetes and its vascular complications [46].
The development of type 2 diabetes, as well as its complications, in particular atherosclerosis, are associated with mtDNA mutations [47][48][49]. Most of the mutations arise because of increased production of ROS near the mitochondrial genome as a result of oxidative stress, which leads to impaired mitochondrial function. In addition, mtDNA mutations are generated by replication errors of mitochondrial DNA polymerase γ and spontaneous base hydrolysis [50]. Recently, a novel m.8561C>G mutation in MT-ATP6/8 (subunits of mitochondrial ATP synthase) has been reported that may be associated with the onset of diabetes mellitus [51]. Another one was demonstrated that nuclear-encoded mitochondrial genes (NEMG) have been identified that code for disease-related proteins that act in key mitochondrial pathways. This may confirm the role of genetic variability in the occurrence of mitochondrial dysfunction, not only as a consequence of DM2, but also as its possible cause [52].
4. Endothelial Dysfunction in Type 2 Diabetes and Its Vascular Complications
The development of vascular complications in type 2 diabetes is associated with toxic effects caused by hyperglycemia, as well as dyslipidemia caused by obesity, gluco-, and lipotoxicity. Chronic hyperglycemia and dyslipidemia lead to increased production of ROS through the activation of various enzymes such as mitochondrial respiratory chain enzymes, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), uncoupled endothelial nitric oxide synthase (eNOS), cyclooxygenase, and xanthine oxidase (XO). ROS-responsive factors increase the production of glyco/lipoxidation end products such as advanced glycation end-product (AGE) and oxidized LDL, which causes endothelial damage and increases intravascular inflammation and leukocyte recruitment, which in turn further increases endothelial dysfunction. ROS production in type 2 DM is increased by accumulation of AGEs and activation of the cellular AGE receptor (RAGE), which promote the secretion of cytokines and stimulate oxidative intermediates under conditions of hyperglycemia [53]. Studies show that AGE/RAGE signaling is involved in diabetes-mediated oxidative stress associated with plaque calcification, endothelial dysfunction, and atherosclerosis progression [54][55]. The damaging effect of ROS is minimized by cellular antioxidant enzymes such as catalase, peroxiredoxins, glutaredoxin (Grx), and glutathione peroxidases (GPx). When these pathophysiological processes are dysregulated, defense mechanisms are reduced, leading to an increase in ROS levels and irreversible damage to key cellular enzymes [56]. Studies show that low antioxidant status predisposes to adverse vascular complications. Thus, a decrease in GPx3 activity is associated with progression of mean carotid intima-media thickness (IMT) and the presence of carotid plaque, which confirms the relationship between GPx3 activity and the pathogenesis of carotid atherosclerosis in patients with type 2 diabetes [57]. It is known that endothelial-cell-derived nitric oxide (NO) plays a protective role in cardiovascular diseases, in particular atherosclerosis, by stimulating vasodilation and inhibiting inflammatory reactions, platelet activation, and aggregation. Violation of the synthesis and/or bioavailability of NO by endothelial cells also leads to endothelial dysfunction. AGEs have been shown to reduce NO production by suppressing the expression of endothelial NO synthase. In addition, oxidative stress caused by AGE-RAGE can also inactivate NO and lead to increased production of peroxynitrite, a toxic by-product of NO. AGE-RAGE stimulates the formation of the endogenous NO synthase inhibitor asymmetric dimethylarginine (ADMA), which also causes endothelial dysfunction [58].
Hyperglycemia and dyslipidemia also promote monocyte adhesion to endothelial cells by inhibiting nitric oxide production and increasing levels of endothelin-1, E-selectin, intercellular adhesion molecule 1 (ICAM-1) and VCAM-1, ROS, angiotensin II, and a plasminogen activator inhibitor [59]. Further, monocytes penetrate into the subendothelial space, differentiating into macrophages that secrete pro-inflammatory cytokines [60]. As a result, LDLs are converted into modified atherogenic LDLs, and oxidation probably occurs in the last stages of modification. These modified LDLs are then taken up by macrophages, leading to foam cell formation and atherogenesis. It is known that AGEs affect the activation of the endothelium and the expression of adhesion molecules, promoting the penetration of monocytes into the subendothelial space, and also enhancing the release of cytokines by macrophages, thereby maintaining the pro-inflammatory effect [61][62]. Glycation of LDL can be considered as a type of the atherogenic LDL modification [63].
It is known that AGEs are formed during periods of hyperglycemia, are poorly metabolized, and slowly accumulate over many years with inadequate glucose control in DM. This is the so-called “metabolic memory”, defined as the long-term influence of the initial glycemic status on the development of diabetic vascular complications, which can accelerate the progression of vascular complications in patients with diabetes mellitus [64].
In addition to being involved in the formation of atherosclerotic lesions, endothelial dysfunction is associated with the development of other diabetic cardiovascular complications of non-ischemic origin. Endothelial-derived cardio-active factors, such as NO, endothelin-1, neuregulin-1 (NRG-1), angiotensin II, prostaglandins, and others, regulate cardiomyocyte activity [65]. In diabetic conditions, endothelial dysfunction leads to disturbed cardiomyocyte metabolism and microvascular coronary disorder, which are the major mechanisms of diabetic cardiomyopathy [66]. The deposition of microvascular AGEs in myocardium causes vascular inflammation and inhibits NO production, ROS production in cardiomyocytes by NADPH oxidase, increased connective tissue crosslinking, and fibrosis development, resulting in predisposition to left ventricular remodeling and diastolic dysfunction [66][67]. Recent meta-analysis demonstrates the association of myocardial fibrosis with DM in clinical studies due to impaired glycemic control, but further investigation of this relationship mechanisms is needed [68]. In addition, endothelial dysfunction in conjunction with increased activation of the renin–angiotensin–aldosterone system and immune dysregulation underlie the pathophysiology of arterial hypertension, one of the major comorbidities of diabetes [69]. Thus, diabetic mechanisms cause heart failure development due to the direct role in cardiac metabolism dysregulation and indirectly through arterial hypertension and coronary atherosclerosis [70].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10051168
References
- Williamson, R.T. On the Treatment of Glycosuria and Diabetes Mellitus with Sodium Salicylate. Br. Med. J. 1901, 1, 760–762.
- Reid, J.; Macdougall, A.I.; Andrews, M.M. Aspirin and Diabetes Mellitus. Br. Med. J. 1957, 2, 1071–1074.
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin Resistance Causes Inflammation in Adipose Tissue. J. Clin. Investig. 2018, 128, 1538–1550.
- Johnson, A.M.F.; Olefsky, J.M. The Origins and Drivers of Insulin Resistance. Cell 2013, 152, 673–684.
- Wu, H.; Ballantyne, C.M. Skeletal Muscle Inflammation and Insulin Resistance in Obesity. J. Clin. Investig. 2017, 127, 43–54.
- Castoldi, A.; de Souza, C.N.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2016, 6, 637.
- Drareni, K.; Gautier, J.F.; Venteclef, N.; Alzaid, F. Transcriptional Control of Macrophage Polarisation in Type 2 Diabetes. Semin. Immunopathol. 2019, 41, 515–529.
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13.
- Ozawa, K.; Miyazaki, M.; Matsuhisa, M.; Takano, K.; Nakatani, Y.; Hatazaki, M.; Tamatani, T.; Yamagata, K.; Miyagawa, J.I.; Kitao, Y.; et al. The Endoplasmic Reticulum Chaperone Improves Insulin Resistance in Type 2 Diabetes. Diabetes 2005, 54, 657–663.
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A Guide to Understanding Endoplasmic Reticulum Stress in Metabolic Disorders. Mol. Metab. 2021, 47, 101169.
- Lin, Y.; Berg, A.H.; Iyengar, P.; Lam, T.K.T.; Giacca, A.; Combs, T.P.; Rajala, M.W.; Du, X.; Rollman, B.; Li, W.; et al. The Hyperglycemia-Induced Inflammatory Response in Adipocytes: The Role of Reactive Oxygen Species. J. Biol. Chem. 2005, 280, 4617–4626.
- Kirichenko, T.V.; Markina, Y.V.; Sukhorukov, V.N.; Khotina, V.A.; Wu, W.K.; Orekhov, A.N. A Novel Insight at Atherogenesis: The Role of Microbiome. Front. Cell Dev. Biol. 2020, 8, 586189.
- Tanti, J.F.; Grémeaux, T.; van Obberghen, E.; le Marchand-Brustel, Y. Serine/Threonine Phosphorylation of Insulin Receptor Substrate 1 Modulates Insulin Receptor Signaling. J. Biol. Chem. 1994, 269, 6051–6057.
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and Cellular Properties of Insulin Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31.
- Takeda, K.; Akira, S. Toll-like Receptors in Innate Immunity. Int. Immunol. 2005, 17, 1–14.
- Yaglova, N.V.; Obernikhin, S.S.; Yaglov, V.V.; Nazimova, S.V. Role of Skin Dendritic and Mast Cells Communications in Triggering Immune Reactions. Clin. Exp. Morphol. 2021, 10, 5–10.
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in Inflammation and Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 85–97.
- Taylor, E.B. The Complex Role of Adipokines in Obesity, Inflammation, and Autoimmunity. Clin. Sci. 2021, 135, 731–752.
- Leal, V.d.O.; Mafra, D. Adipokines in Obesity. Clin. Chim. Acta 2013, 419, 87–94.
- Kraakman, M.J.; Murphy, A.J.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage Polarization in Obesity and Type 2 Diabetes: Weighing down Our Understanding of Macrophage Function? Front. Immunol. 2014, 5, 470.
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and Insulin Resistance. J. Clin. Investig. 2006, 116, 1793–1801.
- Herck, M.A.V.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; Winter, B.Y.D.; Francque, S.M.; Vonghia, L. The Differential Roles of T Cells in Non-Alcoholic Fatty Liver Disease and Obesity. Front. Immunol. 2019, 10, 82.
- Vinué, Á.; Herrero-Cervera, A.; González-Navarro, H. Understanding the Impact of Dietary Cholesterol on Chronic Metabolic Diseases through Studies in Rodent Models. Nutrients 2018, 10, 939.
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344.
- Vonghia, L.; van Herck, M.A.; Weyler, J.; Francque, S. Targeting Myeloid-Derived Cells: New Frontiers in the Treatment of Non-Alcoholic and Alcoholic Liver Disease. Front. Immunol. 2019, 10, 563.
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Investig. 2003, 112, 1796–1808.
- Boutens, L.; Stienstra, R. Adipose Tissue Macrophages: Going off Track during Obesity. Diabetologia 2016, 59, 879–894.
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 Links Innate Immunity and Fatty Acid-Induced Insulin Resistance. J. Clin. Investig. 2006, 116, 3015–3025.
- Shin, K.C.; Hwang, I.; Choe, S.S.; Park, J.; Ji, Y.; Kim, J.I.; Lee, G.Y.; Choi, S.H.; Ching, J.; Kovalik, J.P.; et al. Macrophage VLDLR Mediates Obesity-Induced Insulin Resistance with Adipose Tissue Inflammation. Nat. Commun. 2017, 8, 1087.
- Wunderlich, C.M.; Hövelmeyer, N.; Wunderlich, F.T. Mechanisms of Chronic JAK-STAT3-SOCS3 Signaling in Obesity. JAKSTAT 2013, 2, e23878.
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-Mediated Inhibition of Insulin Receptor Tyrosine Kinase Activity in TNF-Alpha- and Obesity-Induced Insulin Resistance. Science 1996, 271, 665–668.
- Saltiel, A.R.; Pessin, J.E. Insulin Signaling Pathways in Time and Space. Trends Cell Biol. 2002, 12, 65–71.
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in Insulin Receptor Substrate-1 Blocks Interactions with the Insulin Receptor and Inhibits Insulin Action. J. Biol. Chem. 2002, 277, 1531–1537.
- Jager, J.; Grémeaux, T.; Cormont, M.; le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1beta-Induced Insulin Resistance in Adipocytes through down-Regulation of Insulin Receptor Substrate-1 Expression. Endocrinology 2007, 148, 241.
- Simsek, S.; van den Oever, I.A.M.; Raterman, H.G.; Nurmohamed, M.T. Endothelial Dysfunction, Inflammation, and Apoptosis in Diabetes Mellitus. Mediat. Inflamm. 2010, 2010, 15.
- Pangare, M.; Makino, A. Mitochondrial Function in Vascular Endothelial Cell in Diabetes. J. Smooth Muscle Res. 2012, 48, 1–26.
- Tang, X.; Luo, Y.X.; Chen, H.Z.; Liu, D.P. Mitochondria, Endothelial Cell Function, and Vascular Diseases. Front. Physiol. 2014, 5, 175.
- Salnikova, D.; Orekhova, V.; Grechko, A.; Starodubova, A.; Bezsonov, E.; Popkova, T.; Orekhov, A. Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8990.
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. From Mitochondria to Atherosclerosis: The Inflammation Path. Biomedicines 2021, 9, 258.
- Markin, A.M.; Khotina, V.A.; Zabudskaya, X.G.; Bogatyreva, A.I.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis. Life 2021, 11, 165.
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procopio, J.; Morgan, D.; Oliveira-Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes Associated Cell Stress and Dysfunction: Role of Mitochondrial and Non-Mitochondrial ROS Production and Activity. J. Physiol. 2007, 583, 9–24.
- Newsholme, P.; Gaudel, C.; Krause, M. Mitochondria and Diabetes. An Intriguing Pathogenetic Role. Adv. Exp. Med. Biol. 2012, 942, 235–247.
- Ngo, J.; Osto, C.; Villalobos, F.; Shirihai, O.S. Mitochondrial Heterogeneity in Metabolic Diseases. Biology 2021, 10, 927.
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial Dynamics in Type 2 Diabetes: Pathophysiological Implications. Redox Biol. 2017, 11, 637–645.
- Wada, J.; Nakatsuka, A. Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes. Acta Med. Okayama 2016, 70, 151–158.
- Li, M.; Zhu, Y.; Jaiswal, S.K.; Liu, N.F. Mitochondria Homeostasis and Vascular Medial Calcification. Calcif. Tissue Int. 2021, 109, 113–120.
- Permana Maksum, I.; Saputra, S.R.; Indrayati, N.; Yusuf, M.; Subroto, T. Bioinformatics Study of m.9053G>A Mutation at the ATP6 Gene in Relation to Type 2 Diabetes Mellitus and Cataract Diseases. Bioinform. Biol. Insights 2017, 11, 1177932217728515.
- Kirichenko, T.V.; Ragino, Y.I.; Voevoda, M.I.; Urazalina, S.J.; Khasanova, Z.B.; Orekhova, V.A.; Sinyov, V.V.; Sazonova, M.A.; Orekhov, A.N.; Sobenin, I.A. Data on Association of Mitochondrial Heteroplasmy with Carotid Intima-Media Thickness in Subjects from Russian and Kazakh Populations. Data Brief 2020, 29, 105136.
- Kirichenko, T.V.; Ryzhkova, A.I.; Sinyov, V.V.; Sazonova, M.D.; Orekhova, V.A.; Karagodin, V.P.; Gerasimova, E.V.; Voevoda, M.I.; Orekhov, A.N.; Ragino, Y.I.; et al. Impact of Mitochondrial DNA Mutations on Carotid Intima-Media Thickness in the Novosibirsk Region. Life 2020, 10, 160.
- Markin, A.M.; Sobenin, I.A.; Grechko, A.V.; Zhang, D.; Orekhov, A.N. Cellular Mechanisms of Human Atherogenesis: Focus on Chronification of Inflammation and Mitochondrial Mutations. Front. Pharm. 2020, 11, 642.
- Kytövuori, L.; Lipponen, J.; Rusanen, H.; Komulainen, T.; Martikainen, M.H.; Majamaa, K. A Novel Mutation m.8561C>G in MT-ATP6/8 Causing a Mitochondrial Syndrome with Ataxia, Peripheral Neuropathy, Diabetes Mellitus, and Hypergonadotropic Hypogonadism. J. Neurol. 2016, 263, 2188–2195.
- Maude, H.; Lau, W.; Maniatis, N.; Andrew, T. New Insights into Mitochondrial Dysfunction at Disease Susceptibility Loci in the Development of Type 2 Diabetes. Front. Endocrinol. 2021, 12, 694893.
- Hasheminasabgorji, E.; Jha, J.C. Dyslipidemia, Diabetes and Atherosclerosis: Role of Inflammation and ROS-Redox-Sensitive Factors. Biomedicines 2021, 9, 1602.
- Wang, Z.Q.; Jing, L.L.; Yan, J.C.; Sun, Z.; Bao, Z.Y.; Shao, C.; Pang, Q.W.; Geng, Y.; Zhang, L.L.; Li, L.H. Role of AGEs in the Progression and Regression of Atherosclerotic Plaques. Glycoconj. J. 2018, 35, 443–450.
- Markina, Y.V.; Gerasimova, E.V.; Markin, A.M.; Glanz, V.Y.; Wu, W.K.; Sobenin, I.A.; Orekhov, A.N. Sialylated Immunoglobulins for the Treatment of Immuno-Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 5472.
- Bubb, K.J.; Drummond, G.R.; Figtree, G.A. New Opportunities for Targeting Redox Dysregulation in Cardiovascular Disease. Cardiovasc. Res. 2020, 116, 532–544.
- Ling, P.; Shan, W.; Zhai, G.; Qiu, C.; Liu, Y.; Xu, Y.; Yang, X. Association between Glutathione Peroxidase-3 Activity and Carotid Atherosclerosis in Patients with Type 2 Diabetes Mellitus. Brain Behav. 2020, 10, e01773.
- Yamagishi, S.; Matsui, T. Role of Hyperglycemia-Induced Advanced Glycation End Product (AGE) Accumulation in Atherosclerosis. Ann. Vasc. Dis. 2018, 11, 253.
- Laakso, M.; Kuusisto, J. Insulin Resistance and Hyperglycaemia in Cardiovascular Disease Development. Nat. Rev. Endocrinol. 2014, 10, 293–302.
- Lin, P.; Ji, H.H.; Li, Y.J.; Guo, S.D. Macrophage Plasticity and Atherosclerosis Therapy. Front. Mol. Biosci. 2021, 8, 679797.
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus-Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835.
- Markin, A.M.; Markina, Y.V.; Sukhorukov, V.N.; Khaylov, A.M.; Orekhov, A.N. The Role of Physical Activity in the Development of Atherosclerotic Lesions of the Vascular Wall. Clin. Exp. Morphol. 2019, 8, 25–31.
- Aberdeen, H.; Battles, K.; Taylor, A.; Garner-Donald, J.; Davis-Wilson, A.; Rogers, B.T.; Cavalier, C.; Williams, E.D. The Aging Vasculature: Glucose Tolerance, Hypoglycemia and the Role of the Serum Response Factor. J. Cardiovasc. Dev. Dis. 2021, 8, 58.
- Luna, P.; Guarner, V.; Farías, J.M.; Hernández-Pacheco, G.; Martínez, M. Importance of Metabolic Memory in the Development of Vascular Complications in Diabetic Patients. J. Cardiothorac. Vasc. Anesth. 2016, 30, 1369–1378.
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial Cell-Cardiomyocyte Crosstalk in Heart Development and Disease. J. Physiol. 2020, 598, 2923–2939.
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial Dysfunction and Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 851941.
- Horton, W.B.; Barrett, E.J. Microvascular Dysfunction in Diabetes Mellitus and Cardiometabolic Disease. Endocr. Rev. 2021, 42, 29–55.
- Salvador, D.B.; Gamba, M.R.; Gonzalez-Jaramillo, N.; Gonzalez-Jaramillo, V.; Raguindin, P.F.N.; Minder, B.; Gräni, C.; Wilhelm, M.; Stettler, C.; Doria, A.; et al. Diabetes and Myocardial Fibrosis: A Systematic Review and Meta-Analysis. JACC Cardiovasc. Imaging 2022, 15, 796–808.
- Sinha, S.; Haque, M. Insulin Resistance Is Cheerfully Hitched with Hypertension. Life 2022, 12, 564.
- Palazzuoli, A.; Iacoviello, M. Diabetes Leading to Heart Failure and Heart Failure Leading to Diabetes: Epidemiological and Clinical Evidence. Heart Fail. Rev. 2022, 27, 1–12.
This entry is offline, you can click here to edit this entry!