1. Role of CIDE Proteins in Apoptosis
Originally, the CIDE proteins were discovered as apoptotic proteins through a homology search targeting the CIDE-N domain of DNA fragmentation factor 45 (DFF45). The apoptotic pathway is highly regulated and the prevention of its normal activation is linked to several disease states, including cancer [
14]. Near the end of the apoptotic pathway, DNA is condensed and then fragmented by the nuclease DNA fragmentation factor 40 (DFF40). However, DFF40 is maintained as a complex with DFF45, which inhibits DFF40 until it is cleaved by upstream-activated caspases. The interaction between DFF40 and DFF45 occurs at the N-terminal of both proteins, termed the CIDE-N domain. This domain was utilized in a homology search and other CIDE-N domain-containing proteins were identified [
1]. FSP27 (Fat-specific protein, 27 kDa) was technically the first of the CIDE proteins to be discovered by characterization of cDNA clones derived from highly-expressed genes during adipocyte differentiation [
15]. However, it was not until the identification of CIDEA and CIDEB, via homology to the CIDE-N domain, that the alternative names “CIDEC” or “CIDE-3” were used when referring to FSP27 [
1,
16]. Each of the CIDE proteins share homology with the N-terminal of DFF45 to varying degrees (CIDEA, 39%; CIDEB, 29%; FSP27, 38%) and interact with DFF45 at this conserved domain [
1,
2]. DFF45 inhibits the apoptotic action of the CIDE proteins through this domain in a manner similar to its interaction with DFF40.
The apoptotic role of CIDE proteins has been primarily demonstrated through ectopic overexpression in mammalian cells or through induced mutations to different amino acid residues of the individual CIDE proteins [
1,
16,
17]. Initially, the apoptotic functions of the CIDE proteins were thought to be independent of caspase activation [
1]; however, further studies revealed that this function is dependent on caspase-3, caspase-9, and the release of cytochrome c [
18,
19]. In mice, FSP27 forms a homodimer through interactions at the CIDE-N and CIDE-C domains; additionally, mouse FSP27 forms a heterodimer with CIDEA but this interaction occurs at the CIDE-C domain only [
2,
19]. Recently, CIDE-domain containing proteins (Drep2, Drep4, DFF40, FSP27) were discovered to form head-to-tail helical oligomers, a confirmation that is important for their function [
20]. In line with the above observations, both the rate of apoptosis and the level of CIDEA expression were markedly increased in skeletal muscle following an induced burn injury in mice [
21], the CIDE-C domain of CIDEB interacts with the hepatitis C non-structural protein 2 (NS2) in the liver to prevent apoptosis [
18], and the region of the human genome containing FSP27 (chromosome 3p25) was mutated or absent in several forms of human cancers [
16].
For the CIDE proteins to induce apoptosis and fragment DNA, they must localize to the mitochondria [
16,
17]. This notion was challenged by Puri et al. when they identified that FSP27 and CIDEA are highly expressed in adipocytes [
5,
22] where they are localized to lipid droplets. They found that these proteins were not localized to mitochondria, rather they were associated with lipid droplets and that the endogenous proteins have a crucial role in lipid metabolism [
5,
22,
23]. Thus, these proteins were recognized as lipid droplet associated proteins.
A set of elegant studies were performed by Cynthia Smas’ group, in which they discovered a dual role for FSP27 in adipocyte metabolism and cell death. FSP27 is highly upregulated during adipogenesis and is dependent on the differentiation of preadipocytes; the FSP27 transcript is downregulated by the presence of tumor necrosis factor-α (TNF-α), but it is upregulated by the presence of insulin [
24]. Additionally, this group confirmed that FSP27 did not localize to the mitochondria of COS cells. They overexpressed mouse FSP27 ectopically in 293T and 3T3-L1 cells, which resulted in a cellular morphology indicative of apoptosis; however, there was a lack of DNA fragmentation in 3T3-L1 cells, suggesting that the presence of endogenous lipid droplet machinery inhibits the apoptotic action of FSP27 [
24]. Furthermore, in HeLa cells treated with oleic acid, ectopic FSP27 promoted the formation of lipid droplets but did not induce apoptosis [
19].
Overall, the role of CIDE proteins in terms of apoptosis has not yet been fully elucidated, but several required factors have been identified. Taken together, these studies indicate that the CIDE proteins are involved in the DNA fragmentation step of the apoptotic cascade; however, there remains much to be discovered in this area of research. Since their discovery as lipid droplet-associated proteins, much of the work regarding the CIDE proteins has been shifted towards investigating their roles in lipid metabolism.
2. Role of CIDE Proteins in Human Metabolic Health
Gene association studies (also known as polymorphism studies) focus on variations amongst specific genes and alleles and their association with disease. According to the National Center for Biotechnology Information (NCBI), around 6658 single nucleotide polymorphisms (SNPs) have been found in the human CIDEA genes, although not all of them are associated with the disease phenotype. Worldwide clinical studies report around 66 SNPs in CIDEA that are associated with human diseases (NCBI ClinVar). Of these 66 SNPs, 24 are deletions, whereas 42 are duplications.
The human CIDEA gene is located on chromosome 18 (18p11) and spans around 23.22 kilobases (kb) in length, with 4 introns and 5 exons. Linkage studies performed to assess the association of body mass index reported the association of this locus with the development of obesity [
66]. Another independent study performed by Parker et al. determined that the chromosome 18p11 region is positively associated with type 2 diabetes and obesity [
67].
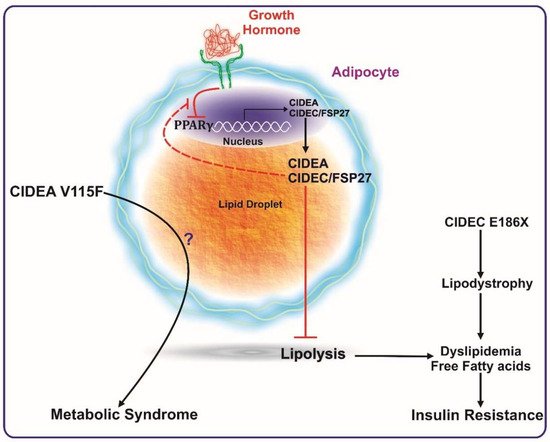
A study by Dahlman et al. revealed the association of a CIDEA polymorphism (V115F) with the occurrence of obesity (see
Figure 1) [
68]. This study was performed in two different Swedish cohorts comprising men and women categorized into non-obese and obese populations. Characterization of the V115F polymorphism revealed six different polymorphic sites in obese individuals, as compared to the non-obese counterparts. However, only one SNP (C19878G→T) was found in the coding region and was responsible for creating the V115F mutation. The study concluded that this polymorphism was positively and significantly associated with obesity and reduced basal metabolic rate. An additional study comprised of 272 male Japanese individuals identified the same polymorphism (V115F) [
69]. This study assessed the association of this amino acid substitution) with the prevalence of metabolic syndrome between two groups—VV vs. VF + FF (valine, V; phenylanaline, F). They concluded that VF + FF individuals have higher abdominal obesity, high fasting plasma glucose levels, and an increased risk of metabolic syndrome. Similarly, an independent study in a Chinese cohort clarified the positive association of the CIDEA V115F polymorphism with the risk of metabolic syndrome [
8].
Figure 1. CIDEA and CIDEC/FSP27 are transcriptionally activated by PPARγ expression. CIDEA and FSP27 bind to lipid droplets and inhibit lipolysis. Reduction in PPARγ activation (such as by growth hormone) reduces the expression levels of these proteins, which ultimately causes enhanced lipolysis, leading to a higher concentration of systemic free fatty acids. The increase in serum free fatty acids culminates in insulin resistance. A mutation in FSP27 (CIDEC E186X) causes lipodystrophy and insulin-resistant type 2 diabetes. The V115F polymorphism in CIDEA is reported to be positively associated with metabolic syndrome. Excess growth hormone levels (such as in acromegaly patients) inactivate PPARγ causing reduced expression of FSP27. FSP27 protects against GH-mediated destabilization of PPARγ to maintain insulin sensitivity.
Another study conducted by Wu et al. studied the association of five polymorphisms in the CIDEA gene with obesity in the Chinese population [
70]. The polymorphisms included were rs1154588/V115F, rs4796955, rs8092502, rs12962340, and rs7230480. Individuals with the rs1154588/V115F, rs4796955, rs8092502, or rs7230480 polymorphism had around 1.4-fold increased risk of developing obesity. Further, the study concluded that individuals with both rs1154588/V115F and rs4796955 polymorphisms were more prone to developing diabetes. In addition to the above SNPs, the association of rs2479 and rs1053239 SNPs in CIDEA with rapid progression of high blood pressure have been reported [
70]. The individuals with rs2479 SNP are more prone to having elevated fasting blood glucose levels along with high triglyceride levels. Overall, the above polymorphism studies unequivocally showed the positive association of CIDEA with healthy metabolic phenotypes in humans.
Functionally, CIDEA was identified as a lipid droplet-associated protein, and its expression was in consonance with insulin sensitivity in obese humans [
7]. In this study, Puri et al. confirmed the PPARγ-mediated regulation of CIDEA expression in white adipose tissue samples from obese individuals undergoing gastric-bypass surgery (see
Figure 1). CIDEA expression in white adipose tissue of obese human subjects was determined and found to correlate positively with whole-body insulin sensitivity independent of gender or BMI. Diet-induced weight loss showed a positive correlation of CIDEA and metabolic health [
71,
72]. Interestingly, changes in adipose tissue CIDEA mRNA levels paralleled variations in insulin sensitivity independently of the change in body mass index [
72]. CIDEA was up-regulated in the adipose tissue of individuals with successful long-term insulin resistance relapse and not in adipose tissue of unsuccessful individuals, suggesting a beneficial role of adipose tissue CIDEA in long term glucose homeostasis, independently of weight variation.
Similar to CIDEA, a specific SNP in FSP27 has also been shown to be associated with the causation of metabolic syndrome. The SNP (G→T) occurs at nucleotide position 556 within exon 6 of FSP27 and inserts a premature stop codon (TAA), resulting in a non-sense mutation (E186X) that disrupts the CIDE-C domain. This mutation has been reported to be associated with partial lipodystrophy and insulin resistance in a female patient (see
Figure 1) [
12].
Homeostatic Model Assessment for Insulin Resistance (HOMA-IR; an index for assessment of insulin sensitivity) is positively correlated with FSP27 expression in BMI-matched obese individuals [
7]. The level of FSP27 in adipose tissue reduces with the development of obesity and is negatively correlated with TNF-α [
73]. Complimentarily, FSP27 expression was found to be decreased with increasing BMI, fasting glucose, fasting insulin, and HOMA-IR [
74]. The latter study elegantly showed that bariatric surgery-induced weight loss caused an increase in FSP27 expression in subcutaneous adipose tissue in parallel to adipogenic and mitochondrial genes. Interestingly, PLIN1 levels were also increased. The latter observation was in line with a positive correlation of adipose PLIN1 expression with insulin sensitivity in obese humans [
7]. Various other studies have suggested the role of PLIN1 in metabolic health in humans [
75], indicating a concerted effort of these lipid droplet-associated proteins in maintaining insulin sensitivity.
FSP27 regulates lipid droplet dynamics and lipolysis in adipocytes through regulation of the catalytic capacity [
44], as well as through transcriptional regulation of adipose triglyceride lipase (ATGL), the rate-limiting enzyme in lipolysis [
46]. These studies demonstrated a mutual relationship between FSP27 and ATGL in regulating lipolysis, triglyceride accumulation, and insulin signaling in human adipocytes. As a proof of concept, the former study showed that FSP27 protects human adipocytes from FFA-mediated insulin resistance [
44], thus highlighting the mechanistic role of FSP27 in maintaining insulin sensitivity, potentially by maintaining an optimal balance of energy storage and breakdown.
Recently, the role of FSP27 in human growth hormone (GH)-induced diabetes was elucidated [
76]. GH-induced lipolysis in human subjects is tightly associated with an acute reduction in CIDEC expression in subcutaneous adipose tissue. Furthermore, the study described that GH-induced lipolysis is mediated via Mitogen-activated protein kinase kinase/Extracellular signal-regulated kinase (MEK/ERK) activation, which suppresses PPARγ transcriptional activity (see
Figure 1) with a feedback loop, by which FSP27 protects PPARγ from being phosphorylated at Ser
273. PPARγ Ser
273 phosphorylation has been linked with the development of insulin resistance [
77,
78,
79]. In human adipocytes exposed to GH, exogenous FSP27 stabilized PPARγ in the nucleus by inhibiting its phosphorylation at Ser
273 [
76]. Another complimentary study in mice and mouse cell lines showed that GH modulation of FSP27 expression is mediated through activation of both MEK/ERK and STAT5 dependent intracellular signaling, whereby, GH-induced MEK/ERK pathway has a predominating role in PPARγ inactivation [
80]. Overall, these studies deciphered part of the molecular mechanism by which FSP27 regulates insulin sensitivity.
Taken together, the various mouse models have shown contradictory results on the role of CIDE proteins in whole-body metabolic phenotype. On the contrary, all of the human studies published to date unequivocally show a negative association of CIDEA and FSP27 with the pathogenesis and pathophysiology of metabolic disease in humans (Figure 1), giving prominence to their role in improving the metabolic health of humans.
This entry is adapted from the peer-reviewed paper 10.3390/cells8030238