Breast cancer is the most commonly occurring cancer in women. There were over two-million new cases in world in 2018. It is the second leading cause of death from cancer in western countries. At the molecular level, breast cancer is a heterogeneous disease, which is characterized by high genomic instability evidenced by somatic gene mutations, copy number alterations, and chromosome structural rearrangements. The genomic instability is caused by defects in DNA damage repair, transcription, DNA replication, telomere maintenance and mitotic chromosome segregation. According to molecular features, breast cancers are subdivided in subtypes, according to activation of hormone receptors (estrogen receptor and progesterone receptor), of human epidermal growth factors receptor 2 (HER2), and or BRCA mutations.
- cancer
- breast cancer
- cancer genomics
- biomarkers
1. Introduction
Breast cancer is a dramatically important health problem, and it is one of the main causes of women death. Using the cancer registers of 187 countries it was estimated that the global breast cancer incidence increased from 641,000 cases in 1980 to 1,643,000 cases in 2010, with an annual increase rate of approximately 3% [1]. Breast cancer killed, in these countries, more than 400,000 women in 2010 [1]. These numbers indicate that there is an absolute need to improve our understanding of the cellular and molecular basis of this tumor, to improve its prevention and therapy. In 2019, about 268,000 new cases of invasive breast cancer are expected to be diagnosed in USA, along with about 63,000 new cases of non-invasive breast cancer; approximately 1% of the breast cancers diagnosed in women is expected to be diagnosed in men [2][3]. Breast cancer rates in USA began to decrease in the year 2000, probably in relation with the reduced use of hormone replacement therapy by women [2]. It was estimated that one in eight USA women will display invasive breast cancer over the course of their lifetime [2][3]. A women’s risk of developing breast cancer nearly doubles if she has first-degree relatives who have been diagnosed with breast cancer [2]. About 5–10% of breast cancers can be linked to gene mutations inherited from one’s mother or father, such as BRCA1 or BRCA2 mutations [2]. The peak of breast cancer incidence occurs for women between the ages of 65 and 80 years. However, invasive breast cancer incidence is also frequently observed in young women (<50 years) and its incidence is increasing 0.2% per year [3]. Considerable progresses have been made over the past 50 years in the evaluation and treatment of patients with breast cancer, leading to a nearly 40% decrease in mortality of this disease (due to prevention strategies and the improvement of medical treatment) [2].
2. Mammary Stem Cells
Breast epithelium forms a ductal network that is embedded into an adipose tissue, connecting the nipple through numerous collecting ducts to a complex system of mammary lobes, which are the structures that are responsible for milk production during pregnancy and lactation. The mammary gland undergoes very extensive changes during development after birth, consisting in its glandular expansion during puberty to full tubule-alveolar differentiation during lactation. Two main cellular lineages are present within the mammary gland: (a) luminal cells, forming the internal layer of ducts and alveoli: these cells express hormone receptors (HR, ER/PR) and they are characterized by the expression of a set of cytokeratins, including CK8, 18 and 19; (b) basal myoepithelial cells, capable of contractile activity and localized between luminal cells and the basement membrane: these cells are characterized by the expression of CK5 and 14 and of smooth muscle actin. These cells constitute a branched, bilayered ductal network that undergoes extensive tissue morphogenesis and regeneration during the lifespan and particularly during puberty and lactation. The main function of luminal cells consists in the generation of milk secretory cells during lactation, while the main function of myoepithelial cells consists in ensuring a propulsive force for milk expulsion.
Early studies showed that any portion of an intact murine mammary gland containing epithelium could generate an entire mammary epithelial tree on transplantation into an epithelium-free mammary fat pad. This capacity was ascribed to the presence in the mammary gland of mammary stem cells that are thought to be self-renewing and to reside at the apex of a cellular hierarchy. The development of cell isolation procedures, as well as of in vivo assay into immunodeficient mice (xenografting of human breast cells into the cleared fat pad or under the renal capsule), have greatly contributed to the characterization of human mammary stem cells. These studies have led to the isolation of cells exhibiting the properties of mammary stem cells while using a combination of various cell-surface markers (CD44, CD24, CD29, CD49f, and EpCAM) [4]. The human breast is formed, starting from a flask-like epithelial structure known as the mammary primordium (this structure develops at week 14 of gestation): central and peripheral primary bud cells can be identified at this stage. At a later stage of development, solid cords of epithelial cells (secondary epithelial outgrowths) migrate in the surrounding mesenchyme, starting from growing primary bud. At these stages of development, fetal mammary stem cells control the growth of mammary gland, displaying properties that are different from those observed for adult mammary stem cells. High CD44 and CD49f expression characterizes a basal epithelial compartment containing all the fetal mammary stem cell activity [5]. Interestingly, during late embryogenesis, fetal mammary rudiments are highly enriched in stem cells [5]. Gene expression, transplantation, and in vitro studies predicted the existence of autocrine and paracrine mechanisms in these fetal mammary stem cells, involving ERB and FGF signaling pathways [5]. The gene expression profiles from mammary stem cells and associated fetal stromal cells displayed a significant similarity with the basal-like and HER2+ breast cancer subtypes [5]. Analysis of normal human breast tissue shows a hierarchical organization, involving non-clonogenic luminal cells, and differentiated (EpCAM+CD49f+ALDH-) and undifferentiated (EpCAM+CD49f+ALDH+) luminal progenitors; all of the progenitor populations are highly plastic and can generate all mammary cell types [6].
In mouse, luminal mammary cells can be subdivided into ER+ and ER-. The cells represent two distinct lineages and their development, homeostasis, and regeneration is distinct and is supported by two different stem cell populations in the adult mammary gland [7][8].
Two different models have been proposed to explain the relationship and the development of various lineages existing in the mammary epithelium. One model assumes that different ductal and lobular progenitors exist, both of which are capable of giving rise to both basal and luminal cells. A second model proposes that early during development the basal and the luminal cell lineages are completely separated.
At variance of mammary fetal progenitors that are multipotent, two separate populations of unipotent progenitor cells maintain the adult mammary gland [9]. In fact, van Keymeulen et al. have used an inducible genetic lineage strategy, allowing to explore the multi- or uni-potency of mammary progenitors during various stages of development: embryogenesis, after birth, during puberty, and lactation. During embryonic life all of the mammary gland cells were derived from a CK14+ multipotent progenitor cell [9]. In contrast, the studies carried out on mammary glands after birth provided evidence regarding the existence of facultative luminal (CK8+) and basal/myoepithelial (CK14+) unipotent progenitors. The luminal progenitors are able to generate mature ductal luminal cells or mature alveolar (milk-secreting) luminal cells; the unipotent basal/myoepithelial progenitors generate myoepithelial cells. However, under appropriate conditions, the adult CD24+CD29+(high) fraction was able to revert to a multipotent embryonic-like activity, generating both luminal and basal cells [9]. Therefore, it was concluded that, in normal adult mammary gland, luminal and basal/myoepithelial lineages both contain long-lived unipotent stem cells displaying extensive renewing capacities, as shown by their ability to clonally expand during morphogenesis and to undergo massive expansion during pregnancy.
Other studies have shown that adult mammary glands contain a Wnt-responsive cell population that is enriched for stem cells. Using a combination of cell culture and in vivo transplantation experiments, it was shown that Wnt proteins act as important self-renewal factors for mammary gland stem cells [10]. In addition, it was shown that Axin 2+ cells (a target of Wnt signaling) have the properties of mammary stem cells [10]. Taking advantage of the identification of Axin2+ cells as a mammary stem cell responsive to Wnt, cell tracking experiments have been carried out to define the staminal potential of these cells during ontogenic development [11]. In the embryo, the Axin2+ cells mark the luminal lineage, while after birth these cells become exclusively committed to the basal cell fate [11]; later, in adult life Axin2 marks cells corresponding to multipotent progenitors generating both mammary lineages and unipotent progenitors, generating both mammary lineages and unipotent progenitors, which are committed to each of the two lineages [11]. These observations indicate that dynamic changes of stem/progenitor cells occur in the mammary gland during development [11]. In line with these observations, the protein C receptor, a Wnt target in the mammary gland, marks a unique population of mouse multipotent mammary stem cells [12].
Some studies have provided evidence regarding the existence of bipotent progenitors in adult mammary tissues, but other investigators did not confirm these findings. Thus, through clonal cell-fate mapping studies, evidence was provided for the existence of bipotent mammary stem cells, as well as of distinct long-lived progenitor cells. The cellular dynamics of these cellular elements at various developmental stages support a model in which both stem and progenitor cells drive breast morphogenesis during puberty, whereas bipotent progenitors drive the homeostasis of adult mammary gland [13]. Cai and coworkers identified a quiescent mammary epithelial cell population expressing high levels of Bcl11b (B-Cell Lymphoma/Leukemia 11B, a zinc finger protein), which were located at the interface between luminal and basal cells [14]. The loss of Bcl11b leads to an exhaustion of ductal epithelium and the loss of epithelial cell regenerative capacity; gain- and loss-of-function studies indicate that Bcl11b induces cells to enter the G0 phase [14]. LGR5 is only a marker of bipotent mammary progenitors in embryonic cells, while in adult mammary LGR5-positive cells are restricted to the myoepithelial lineage [15].
Studies carried out in human breast tissue have supported the existence of a hierarchy of stem/progenitor cells: mammary stem cells (Lin-EPCAM-CD49f+), basal progenitor cells (Lin-EPCAMlow/-CD49f+), luminal progenitor cells (Lin-EPCAM+CD49f+) and mature luminal cells (Lin-EPCAM+CD49f-) [16].
Through asymmetric divisions, these stem cells generate a more differentiated cell progeny unable to self-renew. It was commonly believed that the differentiated cell progeny was unable to revert to a stem-like condition. However, experiments carried out on cultures of mammary epithelial cells have reported an unexpected plasticity, in that differentiated mammary epithelial cells are able to convert a stem-like state, according the stochastic process [17]. This conversion occurs in both normal and transformed mammary epithelial cell populations [17].
Recent studies have shown that a hierarchy of mammary stem/progenitors exists within the mammary epithelium and their basic biology, survival, and proliferation is controlled by signals that are generated both locally and systemically. Among these various signals, hormone signaling plays a key role. The development of mammary gland is controlled by the concerted action of both systemic hormones and growth factors. In this context, a key role is played by steroid hormones estrogen and progesterone that are mitogens for mammary epithelial cells. The effects of these two hormones are mediated through two specific nuclear receptors, the estrogen receptor (ER) and the progesterone receptor (PR), respectively. Studies that have been carried out on developing mouse mammary tissue have clarified the physiological role of these two hormone receptors, with the estrogen receptor being involved in the regulation of duct formation and morphogenesis and the progesterone receptor in the regulation of duct branch formation. Furthermore, PR plays a key role during pregnancy, allowing for tertiary side branching and alveologenesis. The effects of estrogen and progesterone on mammary gland are largely mediated through paracrine mechanisms; estrogen and progesterone markedly affect mammary stem cell function. Thus, it was shown that (i) ovarectomy markedly reduces the mammary stem cell pool; (ii) mammary stem cell activity increases in mice that were treated with estrogen plus progesterone; (iii) mammary stem cell pool markedly increases during maximal progesterone levels at the luteal diestrus phase of the mouse; (iv) treatment with aromatase inhibitors decreases the mammary stem cell pool; and, (v) pregnancy determines a marked increase in mammary stem cell pool, through a mechanism, which is mediated by progesterone and involving a paracrine mediator, RANK Ligand [18][19]. A more recent study has defined the role of RANK as a key paracrine mediator of the effects of progesterone on mammary stem cells. In fact, it was shown that progesterone administration markedly increases the levels of RANK Ligand (Receptor Activator of NF-kB Ligand) [20]. The genetic inactivation of the RANK L receptor impairs the effects of progesterone on mammary stem cells [20]. Importantly, the inhibition of the RANK/RANKL system in mammary gland markedly decreases the incidence and delays the onset of progesterone-driven mammary cancer [20]. A hyperpoliferative RANK+ luminal progenitor cell seems to be the cell driver that generates triple-negative breast cancer in women harboring a germline mutation in the BRCA1 gene [21].
The effect of autocrine and paracrine signals on mammary stem cell fate is ultimately mediated by a fine control of gene expression, which is mainly controlled through the coordinated effects of a network of transcription factors. In this context, a regulatory network orchestrated by the transcription factors Slug and Sox9, plays a key role in the determination of the mammary stem cell fate [22]. The inhibition of either Slug or Sox9 determines a block of mammary stem cell activity [22]. On the other hand, the enforced transient expression of exogenous Slug and Sox9 into differentiated luminal cells determines their conversion to long-term repopulating mammary cells [22].
The ErbB family of receptor tyrosine kinase and their ligands are important regulators of mammary gland development. This family consists of four members: HER1/ErbB1/EGFR; HER2/ErbB2/Neu; HER3/ErbB3; and, HER4/ErbB4. ErB TRKs are required for normal breast development, being particularly important being the role of ErbB2 and ErbB3. In fact, the loss of ErbB2 in mammary epithelium delays ductal elongation and disorganizes terminal end buds of the mammary gland; in contrast, loss of ErbB3, whose expression is highest in luminal mammary cells and lowest in basal stem cells, impaired AKT and MAPK kinase signaling in luminal cells, with the consequent loss of luminal cell proliferation and survival: interestingly, the loss of ErbB3 concomitantly induced an expansion of basal cells, thus suggesting that the normal function of this receptor tyrosine kinase is required for maintaining the balance between luminal and basal breast epithelium [23].
A very important problem in the context of the study of normal and malignant mammary stem cells is related to the development of suitable and reproducible in vivo assays to evaluate mammary stemness. Two types of assays have been proposed in this context. Both of these assays were based on the evaluation of the capacity to regenerate mammary gland structures from mammary human epithelial cells transplanted into highly immunodeficient mice. One of the assays is based on the evaluation of the capacity to colonize the precleared mammary fat pad of immunodeficient mice to create a suitable environment before the transplantation in these sites of human mammary epithelial cells [24][25]. An alternative strategy has been developed that is based on suspending human mammary epithelial cells, together with irradiated human fibroblasts in a collagen gel, which is subsequently implanted under the kidney capsule of estrogen- and progesterone-treated NOD/SCID mice [26].
Advances in next generation sequencing and handling procedures of single cells allowed for the possibility of exploring cellular heterogeneity at the single cell level and reconstruct lineage hierarchies while using single-cell RNA sequencing. Single-cell transcriptomic analysis of stem cell state allows for defining ontogenic stages and lineage specification programs occurring in early murine mammary gland development [27]. This research showed that: (i) individual mammary stem cells co-express genes associated with differentiated mammary lineages; (ii) mammary stem cells constitute a single distribution of heterogeneous transcriptional states, without discrete subpopulations [27]. These findings suggest that stem cell capacity is distributed across heterogeneous cell profiles [27]. Nguyen et al. reached the same conclusions through the study of single cell transcriptomic in human mammary epithelial cell populations, including one basal and two luminal cell types, identified as secretory L1 and hormone-responsive L2-type cells [28]. Temporal reconstruction of differentiation trajectories indicates the existence of one continuous lineage hierarchy that connects the basal lineage to the two differentiated luminal cell branches [28].
Other unicellular sequencing studies have characterized multipotent embryonic mammary progenitors, showing that these cells express a unique hybrid basal and luminal signature and the factors that are associated with the different lineages [29]. Early during embryonic development, embryonic multipotent mammary cells become lineage-restricted [30]. Finally, single-cell landscape in human mammary cells revealed the existence of bipotent-like cells that are associated with breast cancer risk and outcome [31].
3. Molecular Abnormalities of Invasive Breast Cancer
Breast cancer is a highly heterogeneous disease for its histology, epidemiology, and molecular properties. Six molecular subtypes of breast cancer have been identified according to their gene expression profiles and their identification and classification was of fundamental importance for our understanding of tumor genesis and progression: normal breast-like, luminal A and B, basal-like, claudin-low, and HER2/ERB2 overexpressing. The origin of luminal A and B tumors seems to be the mammary duct luminal epithelium, with consistent hormone receptor expression. Basal-like cancers form a heterogeneous group of breast cancers, which probably arise from progenitor cells different from those involved in other breast cancers. HER2/ERB2 overexpressing breast cancers represent a group of aggressive breast cancers that are associated with poor prognosis. Finally, claudin-low are a peculiar group of aggressive breast cancers that are characterized by negative expression of ER, PR, and HER2 (triple-negative), and by the acquisition of mesenchymal/sarcomatoid and/or squamous metaplasia of malignant breast epithelium. This classification also reflects a different metastatic potential of these various breast cancer types. Bone is the most frequent metastatic site for all breast cancer subtypes, except for basal-like. Luminal A tumors are those with the lowest tendency to metastasize; luminal/HER2 and HER2-positive breast cancers were more metastatic than luminal A cancers, particularly at the level of brain, liver, and lung metastases; the basal-like tumors displayed a higher tendency to metastasize at the level of brain and lung, but a lower tendency at the level of liver and bone; finally, triple-negative tumors metastasize at the level of all sites [32].
Other investigators have grouped and classified breast cancers according to the expression of the important functional markers estrogen receptor (ER), progesterone receptor (PR), and HER2, allowing the identification of tumor subtypes with different outcomes [33]. These markers may be also used to additionally characterize the molecular subtypes: luminal A subtype is defined as ER+ and/or PR+, HER2-; luminal B subtype is defined as ER+ and/or PR+, HER2+; basal-like subtype is defined as ER-, PR-, HER2-; and, HER2 subtype is defined as ER-, PR-, HER2+. Thus, luminal A breast cancers are highly ER+ and PR+, HER2-, have usually low proliferative rates and a low Ki67 index, have a NST (no special type), tubular cribiform or classic lobular histology and have a good prognosis. Luminal B breast cancers can be subdivided into HER2- and HER2+: the HER2- tumors are usually ER+ (lower expression than in luminal A tumors), have high proliferation rates, a high Ki67 index, a micropapillary and lobular pleimorphic histology, and exhibit an intermediate prognosis; luminal B, HER2+ breast cancers are usually ER+, PR+, have a high Ki67 index and an intermediate prognosis. HER2-enriched non-luminal breast cancers have NST histology, a high Ki67 index, an aggressive tumor phenotype, and an intermediate prognosis. Triple-negative breast cancers (TNBCs) largely correspond to basal-like and claudin-low subtypes, have a NST histology or a special histology (metaplastic, adenoid cystic, medullary-like), a high Ki67 index, and a poor prognosis.
These three clinically adopted markers for the classification of primary breast cancers are used to help decisions regarding therapy in the metastatic setting. The ER, PR, and HER status often changes during disease progression; in fact, a recent study that was carried out on a large cohort of patients estimated that at relapse 32%, 41%, and 15% of patients change their ER, PR, and HER2 status, respectively [34]. Importantly, women with ER-positive tumors that changed to ER-negative tumors had a significantly 48% increased risk of death when compared with women with stable ER-positive tumors [34].
The treatment of breast cancer during the last years was based on many of the classification criteria that were previously mentioned. The real impact of some of these parameters was now analyzed through the meta-analysis of many clinical trials reporting data in large numbers of patients, thus allowing for reaching some important conclusions. Concerning the hormonal status, the largely more important parameter is the presence of estrogen receptor on tumor cells. In ER-positive breast cancers, the allocation of five-years of treatment with the estrogen inhibitor tamoxifen significantly reduced (of about a third) disease recurrence and disease-related mortality [35]. On the other hand, the meta-analysis of radiotherapy studies that were carried out on more than 10000 patients showed that radiotherapy to the conserved breast halves the rate at which disease recurs and reduces the breast cancer death by about a sixth [36]. However, this proportional benefit varies considerably between patients with different disease characteristics [36]. The meta-analysis of the survival impact of various neo-adjuvant chemotherapy regimens provided evidence that, generally, chemotherapy reduces of about one-third breast cancer mortality as compared to non-chemotherapy and that anthracycline-based regimens are more efficacious than taxane-based regimens or cyclophosphamide-based regimens. Importantly, the risk reductions that are induced by chemotherapy were affected little by age, nodal status, tumor diameter or differentiation, estrogen receptor status, or tamoxifen use [37].
Comparative genomic array studies allowed for identifying three main types of genomic alterations in breast cancer: (a) tumors with few genetic rearrangements (mainly characterized by gain of chromosome 1q and/or loss of 16q); (b) tumors with complex genetic alterations; and, (c) tumors with packed, high-level implicons. Advances in genome sequencing of breast cancers allowed for identifying the full spectrum of mutations present in a small number of breast cancers. One of the large-scale sequencing studies, which was carried out by Stephens and coworkers [38], provided evidence about the existence of different types of alterations, according to three different patterns: (a) interchromosomal translocations with copy number alterations involving large DNA fragments or whole chromosome arms; (b) complex, interchromosomal translocations involving shorter regions with high-level amplifications; and (c) small, interchromosomal segmental alterations, such as deletions, duplications, and/or inversions, called “mutator phenotype”.
The great surprise deriving from the detailed sequencing studies of breast cancer was the observation that individual tumors were unique, each harboring a large collection of individual, “private” mutations that collectively characterized its genome. However, a recent large-scale screening of DNA mutations that occur in breast cancer identified more than 1700 different genic mutations, but only three of these genes were mutated at high frequencies: PI3KCA (43%), TP53 (15%), and MAP3K1 (9%) [39]. The stratification of these patients according to expression subtypes, showed that TP53 mutation is more frequent in basal-like and HER2-enriched disease, while the PI3KCA mutation is more frequent among luminal A tumors [39]. The occurrence of PIK3CA mutations was explored both in in situ and in invasive breast cancers and the conclusion was reached that its frequency is similar in these two tumors, thus supporting the concept that it is more likely to play a role in breast tumor initiation than in invasive progression [40].
Another recent study confirmed these findings; in fact, Banerji and coworkers reported five genes to be frequently mutated in breast cancer: TP53 and PI3KCA, both in 27% of cases; AKT1 in 6% of cases; MAPK1 in 6% of cases; and, GATA3 in about 4% of patients (both AKT1 and PI3KCA mutations activate the PI3K pathway and are mutually exclusive) [41]. Interestingly, in this study, additional recurrent abnormalities occurring in breast cancer have been discovered, including mutations of the transcription factor CBFB (core-binding-factor beta subunit), which is associated with hemizygous deletions of one allele of RUNX1 (4% of cases) and homozygous deletions of RUNX1 (about 2% of cases); a balanced translocation between the MAGI3 and the AKT3 genes leads to the formation of the MAGI3-AKT3 fusion protein, exhibiting constitutive activation of the AKT kinase (about 3% of all cases and more frequent in triple-negative breast cancers) [41]. Ellis and colleagues have also explored the occurrence of mutations in estrogen receptor-positive breast cancers. They have included in their analysis the genomes of tumors that were derived from patients participating in pre-operative clinical evaluation of their response to aromatase inhibitors. This analysis confirmed the frequent mutation of genes, such as PIK3CA, TP53, GATA3, MAP3K1, RB1, and MLL3, which are known to be mutated in breast cancer and led also to the discovery of rare mutations involving genes, such as RUNX1, MYH9, TBX3, and CBFB. Particularly, PIK3CA mutations were observed in 16% of cases, MAP3K1 mutations in 15.5% of cases, and GATA3 mutations in about 9% of cases. These patients were subdivided into two groups according to their sensitivity to aromatase inhibitor treatment: tumors displaying a high frequency of cells expressing the protein Ki67 are aromatase-resistant. Several interesting findings emerged from this comparative analysis: (a) the TP53 mutations were higher in the aromatase-inhibitor-resistant group (38%) than in the aromatase-inhibitor-sensitive group (16%); (b) TP53 mutations were significantly enriched in luminal B tumors and higher histological grade tumors; (c) MAP3K1 mutations were more frequent in luminal A tumors, in grade 1 tumors, and in tumors with lower Ki67 levels (premature inhibitor-sensitive tumors); (d) alterations in DNA replication and mismatch repair are more frequent in the aromatase-inhibitor-sensitive group; and, (e) the presence of mutant GATA3 correlated with suppression of proliferation upon aromatase inhibitor treatment [42].
The integration of genomic and gene expression studies has recently led to the identification of more breast cancer molecular subtypes. This study was based on the analysis of 2000 breast cancers [43]. In this study, Curtis et al. defined 45 regions of sequence amplification or deletion that deregulate genes that are involved in the pathophysiology of cancer [43]. Among these subtypes, particularly interesting was the identification of the ER-positive subgroup that was composed of 11q13/14 cis-acting luminal tumors that harbor other common alterations. This subgroup is a high-risk subgroup. Various putative driver genes reside in the chromosome region, including CCND1, EMSY, PAK1, and RSF1. Additional subgroups identified using this analysis were represented by two subgroups characterized by paucity of copy number and cis-acting alterations. Both of these subgroups have good prognosis and one of them is represented by luminal A cases and is enriched in histiotypes corresponding to lobular and tubular carcinomas; the other subgroup included both ER-positive and ER-negative cases [43]. In addition, several intermediate prognosis groups were identified, including a 17q23/20q cis-acting luminal B subgroups, an 8p12 cis-acting luminal subgroups, and an 8q cis-acting/20q- amplifies mixed subgroup [43]. An additional subgroup within intermediate prognosis group is characterized by the classical 1qgain/16q loss, representing a common translocation event [43]. Within these intermediate prognosis groups are included also basal-like tumors, characterized by high-genomic instability and typical cis-acting alterations, such as 5 loss/8q gain/10p gain/12p gain [43]. Finally, the ERBB2-amplified cancers represent a separate subgroup with negative prognosis and they are composed of HER2-enriched (ER-negative) cases and luminal (ER-positive) cases [43].
Stephens and coworkers have studied somatic mutations and copy-number variants in 100 breast cancers and observed that the number of somatic mutations varied markedly between individual tumors and discovered nine new cancer genes that were rarely mutated, but that can represent driver mutations. The new cancer genes so identified were AKT2, ARID1B, CASP8, CDKN1B, MAP3K1, NCOR1, SMARCD1, and TBX3 [44]. Interestingly, many of these mutations predict the synthesis of truncated, non-functional proteins, thus suggesting that these genes could act as breast cancer tumor suppressors [44]. In this study, it was also observed a strong correlation between mutation number, age at which cancer was diagnosed and cancer histological grade: particularly, it was observed that certain DNA-base substitutions are clearly associated with the age of the patients in tumors not overexpressing the estrogen receptor (estrogen receptor-negative tumors), but not in tumors overexpressing (estrogen receptor-positive) the estrogen receptor [44].
Three recent studies have provided new important insights into our understanding of the life span of breast cancers and of the mutational processes that occur at the level of cancer genome [45][46][47]. Importantly, these studies have provided a mutation life span in the natural history of development of breast cancer. The whole-genome DNA sequencing, through the interpretation of the results that were obtained using sophisticated algorithms, allowed for proposing an archeological map for the accumulation of point mutations and chromosomal rearrangements occurring during the development of breast cancer. At early time points of cancer development, driver mutations (such as TP53 or PIK3CA mutations or ERBB2 amplifications) occur and frequently lead to subsequent large-scale chromosomal instability. Because of this event of fundamental importance in breast cancer tumorigenesis, a clonal population of tumor cells is established, which is identified as the “most recent common ancestor”. This initial event is followed by a long period of time during which the tumor subclones acquire new mutations, which make tumor cells more malignant, through various genetic processes: (a) gradual accumulation of genetic alterations; (b) catastrophic genetic events known as chromothripsis (a genetic event of recent identification, called chromothripsis, to indicate shattering of chromosomes into pieces: the shattering event is followed by the stitching of genomic fragments into derivative chromosomes) or kataegis (a phenomenon that is responsible for the rapid development of point mutations that cause regional accumulation of alkylation-based damage of cytosines and guanines [45][46][47]. Nik-Zainal et al. observed that the regional hypermutation (kataegis) is common in breast cancer and described five different kataegis mutational signatures in these cancers, seemingly occurring through different mutational events: signature A was characterized by C > T mutations at XpCpG sites; signature B was represented by C > T, C > G and C > A mutations at TpCpX trinucleotides; signature C was characterized by C > T, C > G and also C > A mutations at XpCpG trinucleotides; signature D showed a uniform distribution of the different mutational classes; and, signature E was characterized by C > G mutations, but not C > T mutations al TpCpX trinucleotides [45]. Multiple mutation processes contribute to most of the breast cancers, although, in some cases, one process was dominant [45]. Finally, later, during tumor development, a late rate-limiting step is responsible for the emergence of one subclone that becomes dominant due to its capacity to expeditiously grow and represents a significant part of the tumor mass.
The Cancer Genome Atlas Network recently published a large survey of the mutational background occurring in human breast cancer [48] (Figure 1).
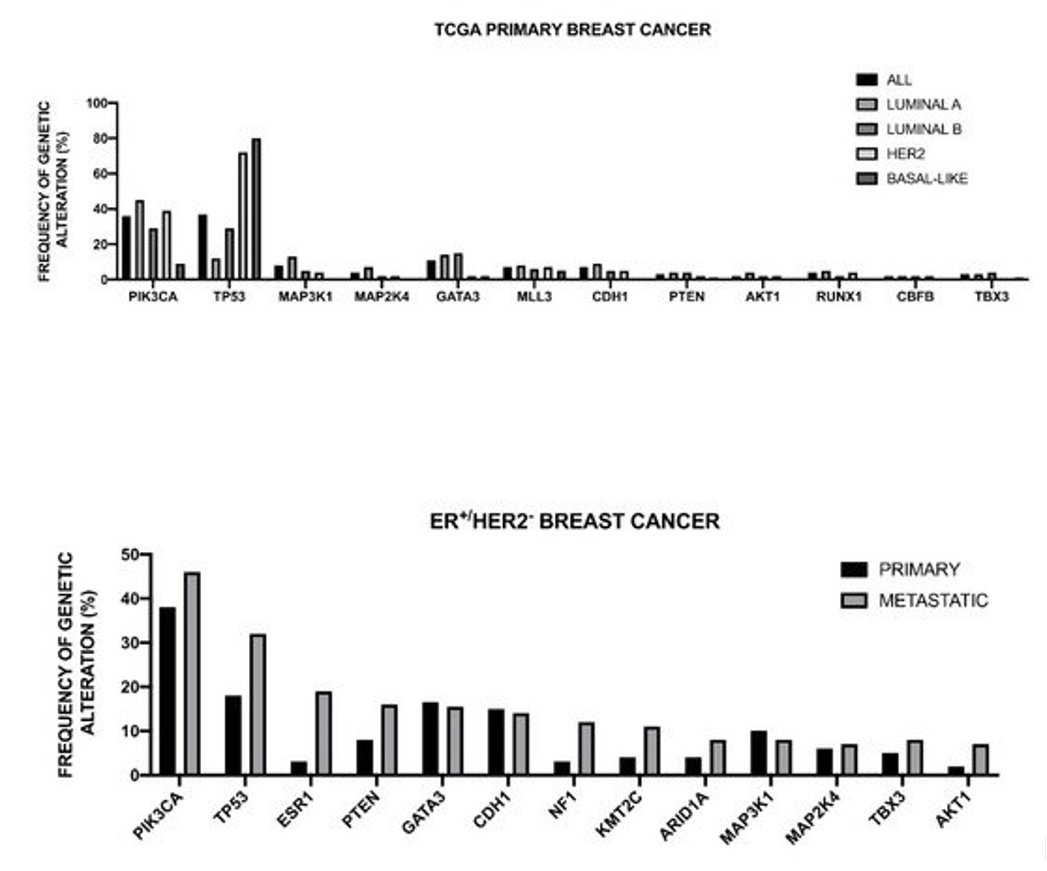
Figure 1. Genomic landscape of recurrent genetic alterations of breast cancer. Top Panel: recurrent genetic alterations in primary breast cancers subdivided according to intrinsic subtype. The data shown in the Figure are reported bt TCGA [48]. Bottom Panel: Most recurrent somatic mutations observed in primary and metastatic ER+/HER2- breast cancers. The data shown in the Figure are reported by Angus et al. [49].
This analysis was, in large part, confirmatory of the results that were obtained in other studies and provided a very useful general overview of the genetics of breast cancers. The luminal A subtype harbored a considerable number of mutated genes, with the most frequent being PI3KCA (about 45%), followed by MAP3K1, GATA3, TP53, CDH1, and MAP2K4. Approximately 12% of these tumors contained inactivating mutations in MAP3K1 and MAP2K4. Luminal B cancers, although, similar to luminal A cancers, exhibited a diversity of significantly mutated genes, with PI3KCA and TP53 (29% of each) being the most frequent [48]. A feature of these tumors was the high mRNA and protein expression of the luminal expression signature, including GATA3 and FOXA1 (mutated in a mutually exclusive fashion), EISR1, XBP1, and MYB (highly expressed, but scarcely mutated). In addition to a higher frequency of TP53 mutations, the luminal B cancers are also characterized for more frequent incidence of p53 pathway-inactivating events, such as ATM loss and MDM2 amplifications. The ensemble of these observations indicates that the TP53 pathway is frequently inactivated in the clinical more aggressive luminal B cancers. The retinoblastoma RB1 expression was detectable in the majority of luminal breast cancers. Cyclin D1 amplification and high expression, common oncogenic events in luminal tumors, are more frequent in luminal B than in luminal A breast cancers.
The HER2 breast cancer subtype was characterized by the frequent amplifications of the HER2 gene (about 80% of cases), frequent TP53 mutations (about 72%) and PIK3CA mutations (39%), and a lower frequency of PIK3R1 (about 4%). Cyclin D1 amplifications were also frequent (about 38%) in these tumors [48]. At the mRNA expression level, a HER2 amplicon signature characterized this group and the expression of EGFR2 and HER2 was observed.
More recently, Nik-Zainal and coworkers reported the landscape of somatic mutations by whole-genome sequencing in 560 primary breast cancers. This analysis showed that at least 12 base substitution mutational signatures and six rearrangement signatures contribute to the somatic mutations found, and 93 mutated cancer genes (31 dominant, 60 recessive, and two uncertain) are involved in the genesis of breast cancer [50]. Three rearrangement signatures, which are characterized by tandem duplications or deletions, are associated with defective homologous-recombination-based DNA repair: one with deficient BRCA1 function, another with deficient BRCA1 or BRCA2 function, and the third related to unknown causes [50]. The global analysis of copy number alterations and somatic mutations generated a total of 1,628 presumptive driver mutations occurring at the level of 93 cancer genes; at least one driver mutation was identifiable in 95% cancers [50]. The ten most frequently mutated genes were TP53, PIK3CA, MYC, CCND1, PTEN, ERBB2, ZNF703/FGFR1 locus, GATA3, RB1, and MP3K1 [50]. TP53, PTEN, and RB1 were more frequently mutated among ER- tumors, while GATA3, CCND1, PI3KCA, ZNF703/FGFR1, MAP3K1, MAP2K4, and CDH1 were more frequently mutated in ER+ tumors [50]. Kataegis (focal base-substitution hypermutation) was observed in 49% of breast cancers [50].
Pereira et al. have reported the analysis of the sequencing of 173 genes that were selected for their “involvement” in breast cancer according to previous studies, in a very large population of 2,433 primary tumors [51]. The mutational landscape was dominated by PIK3CA mutations that occur in about 40% of samples and TP53 mutations occurring in about 35% of cases; the other five genes more recurrently mutated are represented by MUC16 (16.8%), AHNAK2 (16.2%), SYNE1 (12.0%), KMT2C (11.4%), and GATA3 (11.1%) [51]. Some genetic alterations, such as those occurring at the level of PI3KCA, MAP3K1, CDH1, and GATA3 genes are more frequent among ER+ than ER- breast cancers; in contrast, TP53 alterations are markedly more frequent in ER- than ER+ breast cancers [51]. The analysis of gene alterations in histological subtypes showed very remarkable differences; (a) in ductal/NST histotype (largely the most frequent), TP53 and PI3KCA mutations are largely the most frequent; (b) in mixed histotype, PI3KCA mutations are predominant genetic alterations; (c) in mixed histotype, CDH1 and PI3KCA are the most frequent genetic alterations; (d) in the medullary histotype, TP53 is largely the predominant genetic alteration; and, (e) in the mucinous histotype, GATA3 and MAP3K1 are the predominant genetic alterations [51].
Griffith and coworkers have explored the genetic landscape in 1128 primary breast cancer samples [52]. The analysis of the mutation landscape showed that: (i) 17 genes were mutated at a rate greater than 5% and only six at a rate greater than 10%; (ii) the most recurrent mutations were PIK3CA (41.1%), TP53 (15.5%), MLL3 (13.4%), MAP3K1 (12.0%), CDH1 (10.5%), MALAT1 (10%), GATA3 (9.1%), MLL2 (8.7%), ARID1a (7.2%), and BRCA2 (6.6%); (iii) favorable prognostic associations for breast cancer-specific survival were detected for non-silent mutations in MAP3K1, ERBB3, XBP1, and PIK3CA, while adverse prognostic effects were observed for non-silent mutations in DDSR1 and TOP53, as well as for frameshift and nonsense mutations in NF1 [52]. Since PIK3CA and MAP3K1M mutations often co-associate, their combined effect was explored, showing that patients with tumors exhibiting both genes mutated have a more favorable prognosis than cases with either singly mutant gene or without either gene mutated [52].
The PIK3CA gene encodes for the α-isoform of the catalytic subunit (p110α) of PI3K kinase; PIK3CA mutations are observed in 20-40% of early breast cancers and are particularly frequent among hormone receptor-positive tumors. PIK3CA mutations are most frequently localized at the level of three hotspot regions of the PI3KCA protein: p.E542K and p.E545K in exon 10 (corresponding to the helical domain) and p.H1047R in exon 21 (corresponding to the kinase domain) [53]. Several studies have evaluated the prognostic impact of PIK3CA mutations, showing that: (i) in early breast cancer, PIK3CA mutations were significantly associated with a better invasive disease-free survival, distant disease free survival and overall survival [54]; (ii) in ER+/HER2- post-menopausal, early breast cancer BRCA2 mutations and amplifications on 11q13 and 8p11 were significantly associated with increased risk for distant recurrence and PIK3CA mutations were predictive and an improved clinical benefit from letrozole [55][56]. In addition to PIK3CA mutations, gains in PIK3CA copy number (CN) have been observed in breast cancer [57]. Tumors with high PIK3CA CN have more aggressive prognostic features; in ER+/HER2- breast cancers 8-10% of patients display both PIK3CA mutation and PIK3CA CN gain: these patients have a worse outcome, compared to those with PIK3CA mutations without PIK3CA CN gain [57]. In metastatic breast cancer patients the presence of PIK3CA mutations was associated with a reduced survival in HR+/HER2- tumors and with an improved survival in TNBC tumors [53].
In 2013, Ciriello and coworkers performed a comparative analysis of oncogenic signatures among the major human cancers [58]. This analysis showed that tumors are dominated by either mutations (M class) or copy number changes (C class); recurrent chromosomal gains and losses characterizes the C class and includes almost all breast cancers, as well as ovarian cancers [58]. Zack et al. confirmed these findings, who observed that breast cancers, as well as other tumors with frequent CNAs, such as lung cancer, bladder cancer, ovarian cancer, and colorectal cancer, display also with high frequency (45% of cases) whole genome duplication (WGD) [59]. In cancers with WGD, such as breast cancer, most other CNAs occurred after the event of WGD [59].
As previously reported, the acquired CNAs in breast cancers act in cis (a given variant at a locus affects its own expression) or in trans (a given variant affects genes at other sites in the genome) [43]. Approximately 20% of loci exhibit CNA-expression associations in cis and this abnormality includes genes, such as PTEN, ZNF703, MYC, CCND1, MDM2, ERBB2, CCNE1, MDM1, MDM4, CDK3, CDK4, CAMK1D, PI4KB, NCOR1, PPP2RA, MTAP, and MAP2K4 [43]. Trans-acting aberration spots were detected at the level of loci on chromosomes 1q, 7p, 8, 11q, 14q, 16, 17q, and 20q [43].
Single cell genome sequencing methods have been applied to study mutational evolution in ER+ and TNBC patients. Wang et al. have combined this approach with targeted duplex single-molecule sequencing to profile thousands of cells and define the role of some genetic alterations in tumor evolution [60]. The study of two patients (one with ER+ and the other with TNBC) showed: (i) in both patients, a large number of subclonal and de novo mutations gradually evolved over long periods of time, thus generating extensive clonal diversity; and, (ii) in contrast, the single cell copy number profiles were highly comparable, suggesting that chromosome rearrangements occurred early during tumor development, according to punctuated bursts of tumor evolution, leading to stable clonal expansions required for tumor mass formation [60]. Mathematic modeling showed that the TNBC tumor cells displayed an increased mutation rate when compared to ER+ breast cancer cells [60].
Chromosomal instability represents a driving element generating multiple DNA copy-number events, selected during disease development, and resulting in ER+ breast cancers in selection of gene amplification of core regulators of proliferation: in these tumors, stable DNA copy-number amplifications of the core regulators TPX2 and UBE2C are associated with the expression of a whole gene module associated with cell proliferation [61].
Eighty-five percent of the variations in gene expression of breast cancers are due to somatic CNAs at gene loci [43]. Frequently, CNAs involve oncogenes and tumor suppressors that directly affect breast cancer development and progression. Recent studies have measured the existence of a possible association between CNA burden (defined as the percentage of the genome that is affected by CNAs) and tumor grade, recurrence, and metastasis. Thus, Zhang and coworkers have explored the possible role of CNA burden as a prognostic factor associating with the survival outcome of breast cancer patients [62]. In both METABRIC and TCGA data sets, there was an association between CNA burden and patient’s overall survival, in that patients with high CNA burden have a significantly shorter OS than those with low CNA burden [62]. CNA burdens on chromosomes 1, 8, and 16 were significantly higher than other chromosomes: some aberrations on chromosome 1 closely interact with the genes that are involved in the regulation of humoral immune response, while other aberrant genes located on this chromosome belong to MAPK/MAPK3-knockdown related genes involved in cell proliferation; genes that are affected by somatic CNAs on chromosome 8 mainly pertain to the MYC-MAX complex and also include members of the TP53 receptors and ligands gene set; and, genes affected by somatic CNAs on chromosome 16 involve gene sets that are related to cell-cell junction interactions [62]. Furthermore, a string association between CNA burden and age, as well as CNA burden and breast cancer PAM 50 subtypes was observed [62]. Hieronymous and coworkers confirmed these findings, providing evidence that CNA burden of primary and metastatic breast cancers is a prognostic factor, being associated with disease-free and overall survival [63].
Loibl et al. reported the results of a next generation sequencing analysis that was carried out in the neoadjuvant GepurSepto trial; in this study, 851 pretherapeutic formalin-fixed samples were sequenced using a breast cancer-specific hotspot panel of 24 genes (allowing to screen the most relevant mutation and CNA events occurring in this tumor), performing a stratified analysis according to the luminal, HER2-positive, and TNBC subtypes. In this trial, the patients were randomized to either weekly nab-paclitaxel or solvent-based paclitaxel for 12 weeks followed by standard epirubicin/cyclophosphamide; patients with HER2-positive breast cancers received trastuzumab and pertuzumab every three weeks, simultaneously to all chemotherapy cycles [64]. Point mutations and CNAs displayed high heterogeneity among subtypes: TP53 mutations and TP53 and TP2A amplifications were more frequently observed among TNBC; as expected, ERBB2 amplifications are almost exclusively observed among HER2+ tumors; PIK3CA mutations, ZNF703, CCND1, PAK1, and FGFR1 amplifications were more frequently observed among Lum/HER2- breast cancers [64]. In the complete cohort, several genomic alterations were significantly linked to differences in the chemotherapy response in univariate analysis; however, in multivariate analysis, the response to neoadjuvant chemotherapy remained statistically significant for only three of the genomic alterations: PIK3CA mutation, ERBB2 amplification, and PAK1 amplification [64].
4. Molecular Classification of Breast Cancer
Studies that were carried out during the last two decades have strongly supported the prognostic significance and predictive capacity of the breast cancer classification of the four intrinsic subtypes of breast cancer (luminal A, luminal B, HER2-enriched, and basal-like), initially proposed by Perou et al. [65]. These studies started with a genome-wide based ge ne expression profiling from microarray datasets and then moved to a PCR-based test with a curated list of only 50 genes (the so-called “PM-50” gene signature) to classify breast into one of these four groups [66]. The diagnosis by intrinsic subtype improves prognostic and predictive informations to standard a histopathologic parameter for patients with breast cancer [66].
The intrinsic subtypes of breast cancer displayed consistent differences in incidence and response to treatment. The informations that are provided by the intrinsic subtypes complement and expand the information provided by classical clinical parameters and pathologic markers related to the hormonal receptor status. The intrinsic subtype provided prognostic information for patients with metastatic HR+ breast cancer that was treated with first line letrazole ± lapatinib [67]. A recent study explored the changes occurring in intrinsic subtypes at the level of breast cancers undergoing metastatic progression: the rate of subtype conversion was 0% for basal-like tumors, about 23% in basal-like tumors, 30% in luminal B tumors, and 55.3% in luminal A tumors (in large part, these tumors shifted to luminal B) [68].
More recently, the Nanostring nCounter Dx Analysis System provided a system ensuring the more accurate measurements of mRNA expression levels in formalin-fixed tissue when compared to PCR [69]. The system developed while using this system of analysis was called the Prosigna breast cancer signature assay and its prognostic significance was validated [70]. A complete transcript quantification agreement between RNA-Seq and digital multiplexed gene expression platform, and the subtype call after running the PAM assay was observed in a group of breast cancer patients with triple negative cancer [71].
The Prosigna algorithm provides an evaluation of a risk-of-recurrence (ROR) score, being represented as a value from 0 to 100, assessing the risk categories (low, intermediate, or high) and reflecting the 10-year risk of distant recurrence of patients with early-stage HR-positive breast cancer. Many studies have clearly supported the utility of data obtaining the Prosigna assay, combined with standard prognostic criteria, to stratify the recurrence risk [72].
Oncotype DX is one of the earliest clinically validated molecular tests for evaluating the clinical risk in breast cancer patients with early stage disease. This assay was based on the evaluation of 21 genes, of which 16 are tumor-associated and five used as controls; the 16 cancer-related genes include five genes that are involved in proliferation (Ki-67, STK15, Survivin, Cyclin B1, MYBL2), invasion (Stromelysin 3, Cathepsin L2), estrogen (ER, PR, Bcl2, SCUBE2), HER2 (GRB7 and HER2), and GSM1, BAG1, and CD68 [73]. The results of this assay are expressed as a recurrence score (RS): <18RS corresponds to low-risk disease; RS 18–30 corresponds to intermediate-risk disease; ≥31 RS corresponds to high-risk disease [73]. In this assay, a higher expression of genes associated with a favorable outcome (ER, GSTM1, BAG1) is linked to a lower RS, whereas a higher expression of genes associated with an unfavorable outcome, such as Ki67 and cyclin-B1, contributes to higher RS [73]. The Oncotype DX assay was evaluated and validated in the context of the prediction of 10-year recurrence risk in patients with ER+ and LN- breast cancer [73].
The clinical predictive validity of the Oncotype DX assay was evaluated in a large group of postmenopausal women with HR-positive breast cancer that were treated with adjuvant aromatase inhibitors: for patients with node-negative disease, the recurrence rates were 4%, 12%, and 25% for patients with a low, intermediate, and high RS, respectively; for node-positive disease, the recurrence rates were 17%, 28%, and 49% for patients with a low, intermediate, and high RS, respectively [74]. The prognostic value of RS was similar for patients undergoing tamoxifen or aromatase inhibitor treatment [74].
MammaPrint is a 70-gene assay that is based on DNA microarray technology for the assessment of gene expression and quantifies the expression of genes related to tumor progression and metastasis. The FDA approved this test in 2007 for the prediction of the risk of developing metastasis. MammaPrint is currently used in patients with stage II, ER-positive, or ER-negative breast cancers. MammaPrint classifies tumors into groups that are associated with a good prognosis or a poor prognosis on the basis of the risk of recurrence at five years and at 10 years [75]. A large phase III clinical trial explored the clinical utility of MammaPrint as an aid to treatment decisions in early-stage breast cancer. Women at an early-stage of breast cancer were evaluated for the genomic risk while using the MammaPrint assay and for the clinical risk according to standard criteria: at low genomic and clinical risk, these patients did not receive chemotherapy, whereas those with high genomic and clinical risk receive chemotherapy treatment; chemotherapy treatment was used in patients with discordant genomic and clinical risks [76]. The trial that was carried out by Cardoso and coworkers provided evidence that among women with an early-stage breast cancer who were at high clinical risk and low genomic risk for recurrence, the absence of chemotherapy administration on the basis of the MammaPrint assay led to a five-year survival rate without metastasis that was 1.5% points lower than the rate that was observed in patients treated with chemotherapy [76]. A recent study (WSG-PRIMe study) showed that the results of the MammaPrint assay strongly impacted physicians’s therapy decisions in the treatment of patients with luminal early breast cancer [77].
The balance between somatic mutations and alterations in copy number (CNAs) has been investigated in the context of the activities of TCGA, based on the pan-cancer characterization of 12 tumor types [58]. This analysis showed that some of the tumors were dominated by mutations and called M-class tumors, while other tumors are dominated by CNAs and are called C class, such as breast cancer and ovarian cancer [58]. This finding highlights the need for a classification scheme of breast cancers that is based on the pattern of CNAs. To meet this need, Ali and coworkers have performed, in 2014, a large analysis (integration of genomic and transcriptomic profiles) based on over 7500 breast cancer samples and developed a classification system, called IntClus, allowing for the classification of these tumors into 10 IntClust [78]. Integrative cluster 1 is composed by ER-positive tumors, being mainly classified into the luminal B intrinsic subtype; the molecular feature of this cluster is the amplification of the 17q23 locus and GATA3 and TP53 the predominant mutations. Integrative cluster 2 englobes ER-positive tumors and both luminal A and luminal B tumors and it is molecularly characterized by high genomic instability and the amplification of 11q13/14 (involving genes such as CCND1, ERSY, PAK1) and by frequent (about 50%) PIK3CA mutations. Integrative cluster 3 is mainly composed of luminal A subtype and it is enriched for histopathological subtypes, such as invasive lobular and tubular carcinomas, associated with good prognosis; at molecular level, this cluster is characterized by low genomic instability, frequent PIK3CA, CDH1, and RUNX1 mutations, and very rare TP53 mutations. Integrative cluster 4 englobes both ER+ and ER- breast cancers, including 26% of TNBCs and basal-like tumors; at the molecular level, these tumors have low genomic instability and a low level of CNAs; at the mutational level, the most frequent mutations are at the level of PIK3CA (28%) and TP53 (20%). Integrative cluster 5 is composed by both HER2-enriched, ER-negative (58%), and luminal ER-positive tumors, with high-grade tumors with regional lymph nodes involvement; at the molecular level, these tumors are characterized by intermediate levels of genomic instability and a high TP53 mutation frequency (63%). Integrative cluster 6 is a distinct subgroup of ER+ breast cancers, comprising both luminal A and luminal B subgroups and, at the molecular level, is characterized by amplification of the 8p12 locus and high levels of genomic instability; interestingly, these tumors display the lowest PIK3CA mutation rates (about 14%). Integrative cluster 7 mainly comprises ER+ luminal A tumors and corresponds to a good prognostic subgroup; at the molecular level, this cluster is characterized by an intermediate level of genomic instability and specific 16p gain and 16q loss and at mutational level by a high PIK3CA mutation frequency (42%) and by the highest mutation frequency of MP3K1 (32%) and CTCF (11%) among the various clusters. Integrative cluster 8 comprises tumors that are predominantly of luminal A intrinsic subtype, associated with a good prognosis; at the molecular levels, these tumors are characterized by the classical 1q gain/16q loss event corresponding to a common translocation event; these tumors display high levels of PIK3CA, GATA3, and MAPK24 mutations. Integrative cluster 9 is predominantly composed by ER+ tumors of the luminal B subgroup, with an intermediate prognosis; high levels of genomic instability and high mutation frequency of TP53 (58%) and PI3KCA (41%) characterize this cluster. The Integrative Cluster 10 mainly englobes TNBCs and it is molecularly characterized by 5q loss and gains at 8q, 10p, and 12, and by the very frequent TP53 mutations (82%) [79].
A recent study analyzed the relationship existing between the IntClust classification and traditional clinic-pathological features. This analysis showed that: IntClust 3 was enriched for tubular and lobular carcinomas, thus explaining the association with CDH1 mutations in this cluster; mucinous carcinomas were not present in IntClust 5 or 10, but they were scattered in the remaining IntClusts; medullary-like tumors were associated with IntClust 10; HR-positive tumors were scattered along all IntClusts; HER2-positive tumors were predominantly clustered in IntClust 5; and, triple-negative tumors are comprised predominantly in IntClust 10 and in part in IntClust 4 [80].
A statistical framework was recently developed to try to identify peculiar risk groups of breast cancers, while taking the immunohistochemical, intrinsic (PAM50), and integrative (IntClust) subtypes into account. Thus, while using this approach, four late-recurring IntClust subtypes were identified, comprising 26% of ER+/HER2-, each with characteristic genomic copy number driver alterations and with high (42–55%) risk of recurrence up to 20 years post-diagnosis [81]. Furthermore, a subgroup of triple-negative breast cancers that rarely recur after five years and a separate group that remains at risk were identified [81].
The heterogeneity of ER+ breast cancers is supported by many other studies. According to the positivity for PR, ER+ breast cancers can be subdivided into PR+ (more frequent) and PR- (less frequent, 10–155 of all breast cancers, defined as luminal-like). Patients with ER+PR- status exhibit a higher recurrence and worse prognosis, as compared to ER+PR+ tumors. Some studies have explored the molecular features of ER+PR- breast cancers. These tumors have been associated with a significantly higher frequency of HER2 positivity than ER+PR+ tumors. The PR negativity in these tumors might be related to promoter hypermethylation or a loss of heterozygosity at the PR locus. A recent study provided a fundamental analysis of ER+PR- breast cancers, based on the study of five large cohorts of patients. The main results of this study can be summarized, as follows: (i) ER+PR-HER2- tumors displayed lower endocrine responsiveness that did ER+PR+HER2- tumors; (ii) copy number loss or promoter methylation of PR genes occur in about 75% of ER+PR-HER2- tumors, offering an explanation for loss of PR expression; (iii) ER+PR-HER2- tumors displayed higher TP53 (30% vs 17%) and lower PIK3CA mutation rates (25.8% vs 42.7%) and exhibited more ZNF703 (21.5% vs 13.6%) and RPS6KB1 (18.5% vs 7.8%) amplification events than ER+PR+HER2- tumors; and, (iv) ER+PR-HER2- tumors were classified according to the PM-50 gene expression assay as luminal A (46%), but, in part, also luminal B (29%) and basal (16) [82]. It was particularly important to determine the fraction of ER+PR-HER2- tumors that are non-luminal-like in that this subgroup of tumors only showed limited benefit from endocrine therapy, when compared to ER+PR-HER2- luminal-like tumors [82].
Ethier et al. characterized the ER+PR-HER2- subtype, who reached the conclusion that these tumors mainly pertain to the luminal B subtype, and are characterized by higher proliferation and worse outcomes [83]. A systematic review and meta-analysis of the literature data showed that, among patients with hormone receptor-positive breast cancer, patients with either ER+PR- or ER-PR+ tumors have a higher risk of recurrence and shorter survival time than those with ER+PR+ tumors [84]. Patients with both these types of breast cancers need additional or better treatments [84].
Villon-Christersson et al. have reported a cross comparison and prognostic assessment of breast cancer multigene signatures in a large population-based contemporary clinical series of Sweden breast cancer patients. Gene signature classification (the proportion of low- and high-risk) was well aligned with stratification based on current immunohistochemistry-based clinical practice. Most of the signatures did not provide any further risk stratification in TNBC and HER2+ER- patients. Risk classifier agreement in the assessment of ER+ patients was around 50–60%, the disagreement mostly concerning the evaluation of intermediate-risk patients [85]. Most of the investigated gene signatures provided additional prognostic information beyond conventional clinicopathological factors in some specific clinical groups, mainly ER+/HER2- breast cancers [85].
5. HER2 Positive Breast Cancer
Approximately 15–20% of breast cancers are associated with HER2-positivity, being defined as evidence of HER2 protein overexpression that is measured by immunohistochemistry or by fluorescence in-situ hybridization (FISH) measurement of a HER2 gene copy number of six or more [86].
Ross et al, have analyzed 5605 cases of advanced/metastatic breast cancer for HER2 alterations and observed 10.6% of HER2 amplifications and 2.4% of HER2 mutations; 0.7% of cases display co-occurring HER2 amplifications and mutations [87]. Genes commonly co-altered together with HER2 mutations were TP53 (49%), PIK3CA (42%), CDH1 (37%), MYC (17%) and CCND1 (16%) [87]. Kurozumi et al have performed the analysis of HER2 mutational status taking into account all the 1580 cases (including 1301 IDC and 279 ILC) of ER-positive breast cancers included in TCGA, MSK-IMPACT and MTABRIC datasets [88]. ILC tumors accounted for 47.1% of HER2-mutated cases and HER2 mutations were enriched in these tumors (5.7% in ILC vs 1.4% in IDC); HER3 mutations were similarly observed in ILC compared to IDC (1.1% vs 1.8%) [88]. HER2 mutational status was an independent prognostic marker of 10-year overall survival for ILC but not for IDC [88]. Other studies have shown the more frequent HER2 mutations in ILC compared to IDC. Thus Ross et al. showed that 27% of ILCs harbored an HER2 alteration, including in 18% of cases an HER2 mutation, in 5% and HER2 gene fusion and in 5% an HER2 gene amplification [89]. Ding et al reported the characterization of 18 breast cancers with HER2 mutations; HER2 mutations occurred both at the level of tyrosine kinase domain (more frequently) and of the extracellular domain (more rarely); most of the cases with tyrosine kinase domain mutations were ER-positive and had bone metastasis; all the cases with extracellular domain mutations were ER-negative and had no bone metastasis [90]. All the ILC cases displayed a double HER2 mutation.
Yi et al have explored 1184 breast cancer patients with metastatic disease and reported a frequency of HER2 mutations in 8.9% of these patients, with a higher frequency among those with HER2 amplifications compared with those without HER2 amplifications (19.5% vs 4.8%, respectively) [91]. The median progress-free survival of patients with both HER2 amplification and mutation is shorter compared to that of patients with only HER2 amplification following treatment with anti-HER2 monoclonal antibody trastuzumab [91].
Zuo et al. explored the occurrence of HER2 mutations in a cohort of 1248 primary breast cancers and observed a frequency of 2.24% of HER2 mutations; L768S and V773L mutations exhibited a significant increase in tyrosine kinas-specific activity and were observed only in HER2 nonamplified tumors, whereas K753F mutations were found in HER2-amplified tumors; the drug-resistant K753E and L755S HER2 mutations were enriched in metastatic lesions [92].
Mutations in ER represent the most recurrent molecular mechanism involved in the development of resistance to ER-directed therapies, being observed in 25-30% of patients treated with aromatase inhibitors. HER2 mutations may be acquired in ER-positive breast cancer patients under selective pressure of ER-directed therapy based on aromatase inhibitors such as tamoxifen or fulvestrant [93]. HER2 and ER mutations were mutually exclusive [93]. Medford et al. reported the analysis of the genotype profile in circulating tumor cells of 143 patients with endocrine-resistant metastatic breast cancer: 8.4% of these patients displayed HER2 mutations, in 5/12 cases associated with ESR1 co-existing mutations [94].
Recent studies have extensively characterized the genetic heterogeneity of HER2-positive breast cancers. Thus, Ferrari and coworkers have performed a whole-genome sequence and transcriptome analysis of 99 HER2-positive breast cancers, showing that: (i) at gene expression level, four transcriptomic groups were delineated: A and B groups mostly composed of ER+ and PR+ and luminal B tumors, while C and D groups were mostly composed by ER- and PR- and HER2-enriched tumors; (ii) 52 genes were mutated in at least four of these tumors and eight of them are known cancer genes: TP53 (more frequently mutated in ER-than ER+ tumors and particularly in those pertaining to group D), PI3KCA (more frequently mutated in groups A and B), JAK2, ATRX, MAP2K4, ERBB2, KMT2C, and KTM2D; and, (iii) copy number variation affected 59% of the genome and contributed to the molecular heterogeneity of these tumors: gains of 2p and 2q chromosomal arms are more frequent in group D, loss of 11q was more frequent, while the loss of 14q was less frequent in group A, the amplification of CCND1 and PPM1D gene was more frequent in group A [95]. These observations support a consistent heterogeneity of HER2-positive breast cancers.
Daemen and Manning also supported these conclusions, who, through the analysis of published genomic data relative to 3155 breast cancers, reached the conclusion that HER2-positive breast cancers do not constitute a cancer subtype [96]. In fact, HER2 amplification is observed in all breast cancer subtypes, with major characteristics restricted to amplification and the overexpression of HER2 and neighboring genes [96]. Interestingly, HER2-positive tumors are highly enriched in estrogen receptor-driven breast tumors, thus suggesting therapeutic opportunities [96].
Zhao and coworkers have characterized the molecular properties of HER2-positive breast cancers subdivided according to the expression of ER and PR, with particular emphasis on the triple-positive (ER+PR+HER2+) subgroup [97]. According to the hormone receptor expression, HER2-positive breast cancers can be subdivided into ER+PR+HER2+ (TPBC), ER+PR-HER2+, ER-PR+HER2+ (very rare), and ER-PR-HER2+. The triple-positive subgroup displays several peculiarities: (i) it had a significantly better prognosis than the group ER-PR-HER2+; (ii) TPBCs displayed a lower TP53 mutation rate than ER-PR-HER2+ breast cancers (30% vs 69%); and, (iii) TPBCs exhibited lower HER2 mRNA and protein expression than ER-PR+HER2+ tumors [97]. More than 40% of TPBCs can be classified as luminal A and these patients have a better prognosis than those with TPBC of other subtypes [97]. Interestingly, this study also showed that MUC16, GATA3, and ERBB3 mutations are strongly associated with the ER+PR-HER2+ phenotype [97]. Finally, concerning CNVs, TPBCs display less frequent MYC amplification and NCOR2 loss, but more frequent CCND1 amplification and FANCA loss than ER-PR-HER2+ tumors [97].
Chen et al evaluated the genomic profiles of 107 stage I-III Chinese HER2-positive patients, 64 HR+/HER2+ and 43 HR-/HER2+: HR+/HER2+ tumors displayed more gene amplification, splice site and frameshift mutations and a lower number of missense, nonsense and insertion-deletion mutations compared to HR-/HER2+ tumors [98]. An analysis of the altered pathways showed that HR+/HER2+ tumors showed more mutations in genes involved in homologous recombination (37.5% vs 11.6%), TGF-beta (35.9% vs 11.6%) and WNT (45.3% vs 16.3%) [98].
The analysis of primary HER2-positive breast cancers that were obtained from premenopausal Asian women with recurrent breast cancers showed some differences in the rate of several genetic abnormalities, compared to corresponding non-Asian premenopausal breast cancers: particularly, TP53, KTM2D, KMT2C, and SDK1 gene alterations were significantly more frequent in Asian than in non-Asian HER2-positive breast cancer patients [99].
Few studies have explored the genetic abnormalities of HER2-positive breast cancers resistant to trastuzumab therapy. A recent study showed that the mutational burden in heavily treated trastuzumab-resistant HER2-positive metastatic breast cancer is highly variable and not directly correlated with outcome [100]. The activation of the MAPK/ERK pathway through mutations in EGFR, BRAF, or KIT might mediate resistance to trastuzumab [100].
Importantly, the intratumoral heterogeneity of HER2-positive breast cancers with respect to tumor genomics significantly affected the probability of achieving a pathological complete remission following neoadjuvant therapy that is based on chemotherapy and HER2 targeting with specific monoclonal antibodies [101].
Recent studies indicate that a peculiar gene signature could predict the response to therapy of HER2+ breast cancers. This predictive gene signature was obtained while taking two important elements into account: (a) HER2 overexpression drives mammary carcinogenesis, via its effects on normal and malignant mammary stem cells; (b) in a mouse mammary model of HER2+ breast cancer, mammary stem cells (CD24+/JAG1-) have been purified and used to generate a 17-gene signature [102]. This HER2 Tumor Initiating Cells-Enriched Signature (HTICS) consists of eight up-regulated (AURKB, CCNA2, SCRN1, NPY, ATP7B, CHAF1B, CCNB1, CLDN8) and nine downregulated genes (NRP1, CCR2, C1QB, CD74, VEAM1, CD180, ITGB2, CD72, ST8SIA4): the up-regulated set includes genes that are associated with passage through the S/G2/M phase of the cell cycle, whereas the down-regulated genes include genes that are involved in cell adhesion, angiogenesis, and immune-response [102]. This signature was specific for HER2+/ER- breast cancers and it identifies tumors that are resistant to chemotherapy, but sensitive to chemotherapy+trastuzumab [102].
Lesurf et al. provided clear evidence that HER2-positive breast cancers classified as HER2-enriched achieved significantly higher rates of complete remission when compared to those as luminal A, luminal B, or basal-like [103]. Furthermore, immune and inflammatory signatures correlated with response to neoadjuvant therapy based on chemotherapy and anti-HER2 antibody [103]. Several other studies supported these conclusions. HER2-positive breast cancer consists of four intrinsic molecular subtypes, luminal A, luminal B, basal-like, and HER2-enriched, with the last-one being the predominant subtype corresponding to about 60–70% of all HER2-positive breast cancers. HER2-enriched subtype is a predictor of complete response following neoadjuvant therapy with dual HER2 inhibitors (trasuzumab and lapatinib) without chemotherapy in early stage HER2-positive breast cancers [104]. Furthermore, in the randomized clinical trial NeoALTTO, the expression of HER2 and the HER2-enriched subtype were the most significant predictors of pathological response [105]. Finally, Pernas et al. reported the analysis of intrinsic tumor subtypes and residual tumors following neoadjuvant trastuzumab-based chemotherapy in a group of 150 patients with stage II-IIIC HER2-positive breast cancers [106]106]. This study was focused in order to evaluate the association of genomic variables with pathologic response [106]. In these patients, the complete pathological response after neoadjuvant chemotherapy was 53% with higher responders among HR-negative tumors when compared to HR-positive tumors (70% vs 39%); the HR-negative HER2 breast cancers were enriched in HER2-enriched tumors (75%) [106]. The study in pre- and post-treatment samples derived from patients not achieving a complete pathological response, showed a lower proportion of HER2-enriched and twice the number of luminal tumors were observed at baseline, and luminal A was the most frequent intrinsic subtype in residual tumors; interestingly, the majority of luminal A tumors maintained the same subtype in residual tumors, whereas HER2-enriched tumors changed to non-HER2-enriched tumors [106].
In a recent study, Prat and coworkers evaluated 305 breast cancer patients with early HER2-positive disease and 117 patients with advanced HER2-positive disease. These patients were evaluated in the context of five different clinical trials for the response to dual HER2 blockade therapy and HER2-enriched subtype with the PAM 50 assay and ERBB2 mRNA levels [107]. In early disease, the HER2-enriched subtype corresponded to about 84 and 45% of ERBB2-high and ERBB2-low tumors, respectively. After lapatibin and trastuzumab neodjuvant treatment, the HER2-E/ERBB2-high group achieved a rate of complete pathological responses that was higher than the rest of HER2-positive patients (44.5% vs 11.6%); similar findings were observed in early patients undergoing neoadjuvant treatment with trastuzumab and pertuzumab (66.7% complete responses among HER2-E/ERBB2-high patients, when compared to 14.7% in the rest of patients); finally, the HER2-E/ERBB2-high group exhibited longer PFS and OS in patients with advanced disease [107].
HER2-positive breast cancers exhibit aggressive clinical behavior, responding only moderately to chemotherapy, and have higher rates of recurrence and metastasis. The introduction of HER2-targetd therapies, including the monoclonal antibodies anti-HER2 trastuzumab and pertuzumumab and the HER2 tyrosine kinase inhibitor lapatinib, has revolutionized the therapy and substantially improved the outcomes of patients with HER2-positive breast cancers. However, the development of resistance to anti-HER2 treatment represents a consistent challenge, which indicates the clinical need for novel therapies.
Importantly, recent studies have reported the long-term effects of trastuzumab administered alone or in combination with chemotherapy to HER2-positive breast cancer patients, all showing a significant effect of this drug on disease-free survival and overall survival [108][109][110].
In this context, recently, two new drugs targeting HER2 were introduced in the clinical treatment of HER2-positive breast cancers: (i) trastuzumab emtansine, an antibody-drug conjugate of trastuzumab with the cytotoxic agent emtansine, a microtubule inhibitor; (ii) pyrotinib, an irreversible pan-ERB receptor tyrosine kinase inhibitor targeting HER1, HER2, and HER4. Thus, the risk of recurrence of invasive breast cancer or of death was 60% lower with adjuvant trastuzumab emtansine than with trastuzumab alone among patients with HER2-positive early breast cancer who had residual invasive disease after completion of neoadjuvant therapy [111].
Lapatinib combined with capecitabine is one of the recommended regimens for patients with HER2-positive metastatic breast cancer who have been pretreated with taxanes, anthracyclines, and trastuzumab. In this context, a recent study showed that, in women with HER2-positive metastatic breast cancer treated with taxanes, anthracyclines, and/or trastuzumab, pyrotinib plus capecitabine elicited significantly better overall survival than lapatinib plus capecitabine [112].
As repeatedly emphasized, HER2-positive breast cancers are a family of distinct diseases, particularly including ER- and ER+ tumors. HER2-positive tumors display important biological differences, implying differences in drug sensitivity. Data that were derived from a consistent set of clinical trials indicate that, within the group of HER2-positive tumors, the ER- subgroup is clearly more sensitive than the ER+ subgroup to anti-HER2 treatment [113]. More particularly, patients with triple-positive breast cancer are less responsive than patients with HER2-posive, ER-negative tumors to achieve a pathologic complete response in neoadjuvant trials, including HER2-targeted therapies, involving the dual blockade of the HER2 receptor with trastuzumab and pertuzumab or combination treatment with the tyrosine kinase inhibitor klepatinib [113]. In this context, an example of this research is given by the CALGB 40601 clinical trial, a randomized phase III trial evaluating HER2 targeting with paclitaxel plus trastuzumab with or without lapatinib in HER2-positive patients with operable HER2-positive breast cancer [101]. This trial evaluated the capacity of the neoadjuvant treatment to induce pathologic complete response while considering the phenotypic and molecular heterogeneity of HER2-positive breast cancers [101]. The results of this research clearly showed that the rate of responses to treatment were clearly influenced by HR status and the intrinsic subtype; thus, ER+/HER2+ tumors responded less to HER2-targeting treatments than ER-/HER+ tumors; tumors pertaining to the HER2-E subtype respond better than luminal A and luminal B to HER2-targeting therapy [101] (Figure 2). In a subsequent analysis of the pretreatment tumors of these patients by mRNA sequencing and DNA exome sequencing, it was shown that somatic DNA alterations (mutations and DNA copy number alterations), tumor molecular subtype and the microenvironment (immune cells) were independent predictors of response to neoadjuvant treatment [114].
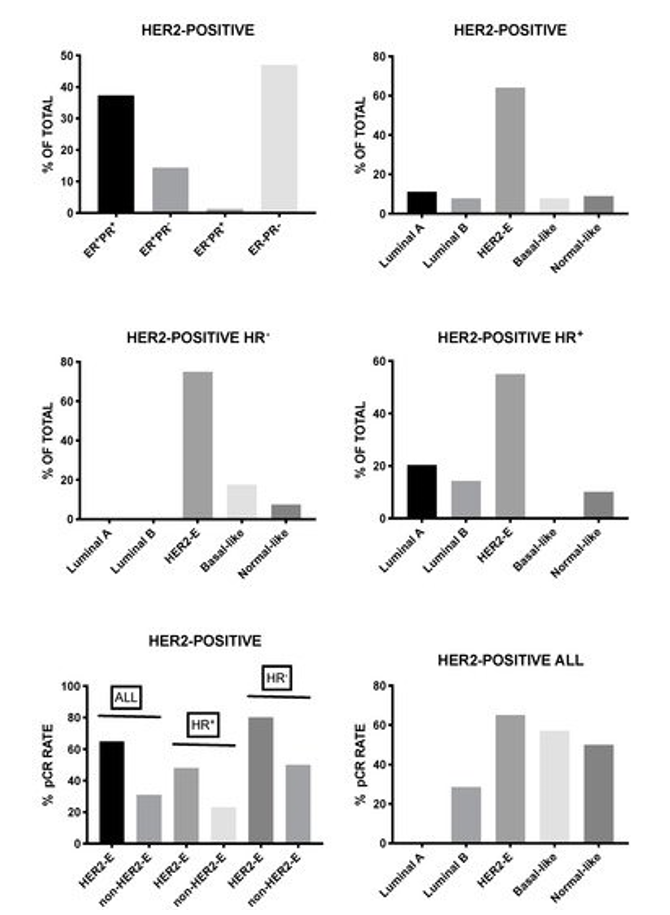
Figure 2. Hormone receptor status (top panel, left) and intrinsic subtype overall (top panel, right) or by hormone receptor status (middle panels) in a population of HER2-positive breast cancer patients undergoing neoadjuvant treatment with trastuzumab plus paclitaxel with or without lapatinib. The bottom panels report the rate of pathological complete response (pCR) observed in all HER2-positive patients (right panel) or in HER2 patients with a HER2-enriched expression subtype, subdivided according to hormonal receptor status (left panel). The data shown in this Figure are reported by Carey et al. [101].
HER2-positive breast cancer patients consist of four intrinsic molecular subtypes, luminal A, luminal B, basal-like, and HER2-enriched, the last one being the predominant subtype corresponding to approximately 60–70% of all HER2-positive breast cancers. HER2-enriched subtype is a predictor of complete response following neoadjuvant therapy with dual HER2 inhibitors (trastuzumab and lapatinib) without chemotherapy in early stage HER2-positive breast cancer [104]. The presence of stromal tumor infiltrating lymphocytes (TILs) is associated with complete response and improved outcomes in HER2+ breast cancer patients treated with trastuzumab plus chemotherapy. The analysis of patients enrolled in the PAMELA trial showed that the number opf TILs during treatment, but not at baseline, is an independent determinant of response to neoadjuvant anti-HER2 therapy [115].
In the randomized clinical trial NeoALTTO, the expression of HER2 and the HER2enriched subtype were the most significant predictors of pathological response [105]. The 41-gene classifier TRAR was able to identify patients sensitive to anti-HER2 enrolled in the context of the NeoALTTRO study [116].
Pernas et al. have reported the analysis of intrinsic tumor subtypes and residual tumors following neoadjuvant trastuzumab-based chemotherapy in a group of 150 patients with stage II-IIIC HER2-positive breast cancers [106]. This study was focused in order to evaluate the association of genomic variables with pathologic response [106]. In these patients, the complete pathological response after neoadjuvant chemotherapy was 53% with higher responders among HR-negative tumors when compared to HR-positive tumors (70% vs 39%); the HR-negative HER2 breast cancers were enriched in HER2-enriched tumors (75%) [106]. The study in pre- and post-treatment samples that were derived from patients not achieving a complete pathological response, showed a lower proportion of HER2-enriched and twice the number of luminal tumors were observed at baseline, and luminal A was the most frequent intrinsic subtype in residual tumors; interestingly, the majority of luminal A tumors maintained the same subtype in residual tumors, whereas the HER2-enriched tumors changed to non-HER2-enriched tumors [106]. A large meta-analysis of literature data showed that the HER2-enriched biomarker identifies breast cancer patients with a higher chance of achieving a complete pathological response following neoadjuvant anti-HER2-based therapy beyond hormonal receptor status and chemotherapy [117].
Preclinical studies have supported the use of CDK4/6 inhibitors in the treatment of HER2-positive breast cancers, this strategy being particularly promising in HER2+ER+ tumors for the presence of a crosstalk between signaling-linked to HER2 and ER. The phase II open label NA-PHER2 trial evaluated the activity of a combination regimen that was based on trastuzumab, pertuzumab, pabociclib, and fuvestrant in patients with triple-positive breast cancers [118]. This regimen elicited a marked reduction of Ki67 expression at week two post-treatment and at surgery after 16 weeks of treatment; importantly, 50% of treated patients achieved a complete clinical response and 27% achieved a pathological complete response [118]. The results of this study support the further exploration of CDK4/6 inhibitors in combination with anti-HER2 therapy and endocrine therapy in the therapy of patients with triple-positive breast cancers. Additionally, preliminary results of the SOLTI-1303 PATRICIA trial further supported the use of CDK 4/6 inhibitors in the treatment of triple-positive breast cancers. The results on the first 45 recruited patients showed that palbociclib in combination with trastuzumab is safe and active in HER2-positive breast cancers with advanced disease, pre-treated with trastuzumab, particularly at the level of HER2+/ER+ patients [119]. Patients HER2-positive pertaining to luminal intrinsic subtypes respond much better than those that correspond to non-luminal subtypes [119].
Tumors displaying HER2-mutated tumors represent a peculiar subgroup of HER2+/ER+ breast cancers. A recent study showed that ERBB2-activating mutations occur with increased frequency in metastatic breast cancers; in fact, 70% of the ERBB2 mutations are detectable in HER2+ER+ non-amplified breast cancers, with a higher frequency occurring in metastatic tumors (4.3%) compared to primary cancers (2.5%) [120]. Importantly, the inhibition of mutant HER2 function with neratinib restores the efficacy of antiestrogen therapy [120]. In line with these findings, it was proposed dual blockade of the HER2 and ER pathways as a strategy that is required for the treatment of HER2+/ER+ breast cancers [120]. Neratinib is a TKI, approved by FDA for extended adjuvant treatment of early-stage HER2-amplified breast cancer following completion one-year of trastuzumab-based therapy. The approval was based on the results of the ExteNET randomized study evaluating neratinib when compared with placebo as an extended adjuvant therapy in patients with early-stage HER2-positive breast cancer who had completed treatment with trastuzumab: patients that were treated with neratinib had significantly fewer iDFS (invasive disease-free survival) events than those did in the placebo group [121]. Importantly, the benefit of adjuvant neratinib was more pronounced in patients with ER+ breast cancers [121]. Some authors suggested the capacity of neratinib to block CCND1 (cyclin D1) and through this mechanism to maintain tumor suppression [122].
Neratinib therapy administered under form of monotherapy or in combination with hormonal deprivation therapy showed a significant clinical benefit in a population of 28 to 46% of HER2-mutated breast cancer patients, associated a median progression-free survival ranging from 3.6 to 5.4 months [123][124][125].
The analysis of the long-term outcomes of HER2-positive patients that were enrolled in the GepartQuinto trial showed a similar survival benefit in patients HR+ receiving prolonged anti-HER2 treatment with neoadjuvant lapatinib, followed by adjuvant trastuzumab [126].
Sudhan explored the mechanisms through which extended adjuvant neratinib treatment achieved a better clinical outcome in patients with HER2+ER+ breast cancers; these authors have developed a human-in-mouse breast cancer model and, through the analysis of this model, reached the conclusion that HER2+/ER+ tumors rapidly evade ER blockade through ERBB pathway hyperactivation and incomplete suppression of cyclin D1 by estrogen inhibitors, requiring the complete suppression of the effect of nerotinib [127].
Unfortunately, the clinical benefit deriving from ceritinib administration to ER+/HER2mut/HER2-non-amplified metastatic breast cancers is limited. The results of a recent phase II trial showed that in patients with ER+/HER2mut/HER2-non-amplified tumors in fulvestrant-treated, fulvestrant-naïve and ER- patients the clinical benefit rate deriving from ceritinib administration was 38%, 30% and 25%, respectively [128]. Both primary and secondary resistance mechanisms limit the benefit deriving from ceritinib administration. Smyth et al. have evaluated 81 HER2-mutant metastatic breast cancer patients undergoing treatment with nerotinib alone or in combination with fulvestrant; progression-free survival and duration of response were longer in ER+ patients receiving combination therapy [129]. Two mechanisms of modulation of the response to ceritinib were identified in these patients: preexistent concurrent activating mutations of HER2 and HER3 were associated with poor treatment outcome; acquisition of multiple HER2 activating events. As well as gatekeeper alterations, were frequently observed in patients deriving clinical benefit from neratinib [124].
Hacker explored the role of co-occurring HER2 and HER3 mutations in mediating HER2 inhibitor sensitivity. The analysis of large cancer datasets on the genetic alterations of cancer patients showed that 8.3% of HER2-mutated cancers have HER3 mutations, while only 2.3% of HER2-WT cancers display HER3 mutations [129]. HER3 and PIK3CA mutations do not co-occur in breast cancer; Mutant HER3 and HER2 form a HER2-HER3 dimer acting as a strong PIK3-AKT activator [130]. Studies in cell lines with HER3/HER2 mutations showed that the drug combination of neratinib and a PI3K alpha inhibitor (alpelisib) resulted in inhibition of cancer cell growth [130]. The results of the SUMMIT clinical trial showed that patients with concurrent HER2 and HER3 mutations had poor treatment responses to neratinib therapy [124].
Smith et al. reported the genomic profiling of 733 HER2-amplified breast cancers and showed in metastatic tumors an enrichment of mutations the promote MEK/ERK signaling, such as HER2 mutations (7% in metastatic versus 3% in non-metastatic tumors) and NF1 mutations (7% in metastatic tumors versus 4% in non-metastatic tumors) [131], These observations suggest that HER2 resistant breast cancers lose AKT dependence and acquire a marked sensitization to MEK/ERK inhibition [131].
Chui et al. explored HER3 expression in invasive breast cancer and observed its overexpression in 10% of cases [132]. In tumors with normal expression levels of HER1 and HER2, the overexpression of HER3 displayed a significant negative prognostic effect on disease-specific overall survival, independent of clinicopathological parameters affecting survival [132].
The study of the interaction of HER2 and HER3 at molecular level is fundamental to better understand the mechanisms of responsiveness/resistance of HER2-mutated breast cancers to HER2 inhibitors. Diwanji and coworkers using cryo-electron microscopy reconstructed the structure of the HER2-HER3 heterodimer bound to the growth factor neuregulin-1β (NRG1β); the introduction in this molecular complex of a mutant HER2 harboring S310F induced a stabilization of the HER3 dimerization arm and increased the total buried surface area between the HER2-HER3 interface [133].
Two recent studies better clarified the HER2-HER3 interaction and the possible targeting of this molecular complex. In these studies, Campbell and coworkers undertake a structure function analysis of HER2-HER3 complex [134][135]. In a first study, these authors showed that receptor conformation and plasticity is very pronounced and the extracellular domains of HER2 and HER3 molecules are not effective targets for blocking HER2-HER3 signaling transactivation [134]. In a second study, they showed that some regions of the HER2-HER3 dimers represents potential molecular targets, such as the HER2 kinase receiver interface and, particularly, the HER3 AP-2 pocket site [135].
A recent study showed the existence of a peculiar mechanism of drug resistance occurring in some HER2-positive breast cancers. This resistance mechanism is related to a sub-population of cancer cells, “drug-tolerant persisters” (DTPs), that survive through irreversible, non-genetically mediated mechanisms [136]. The treatment with HER2-targeting agents induces the transition of a subpopulation of quiescent breast cancer cells from a pre-DTP condition to a DTP state [136]. At biochemical level, this transition involves a rewiring of the PI3K/AKT/mTORC1 pathway to enable AKT-independent mTORC1 activation [136].
Preclinical studies support the clinical use of agents targeting HER3 in HER2-mutant breast cancer cells [137][138]. The anti-HER3 antibody drug conjugate EV20/MMAF displayed potent anti-cancer activity against different models of primary resistance and secondary resistance to anti-HER2 therapies, including trastuzumab, lapatinib, neratinib, and trastuzumab-emtansine [137].
A recent study reported the characterization of a trispecific monoclonal antibody to HER2, CD3 and CD28 that induces regression of breast cancer cells through a mechanism involving CD4-dependent inhibition of tumor cell cycle progression; this antibody may offer a new tool for therapeutic targeting of HER2-positive breast cancers [139].
In spite the occurrence of resistance mechanisms that considerably limit the effectiveness of HER2 targeting, the meta-analysis of randomized clinical trials involving the treatment of early-stage, HER2-positive breast cancer with either chemotherapy plus trastuzumab versus chemotherapy alone showed that adding trastuzumab to chemotherapy significantly reduces recurrence and mortality from breast cancer by a third [140].
6. Genetic Abnormalities of Triple-Negative Breast Cancer
Nearly 10% to 20% of primary breast cancers are triple-negative breast cancers that lack expression of ER, PR and HER2, have usually a high-degree at presentation and display frequent TP53 mutations.
Although often thought to be synonymous, TNBC and BLBC (basal-like breast cancer) represent different biologic phenomena. Rakha and coworkers have explored this important topic by studying two large cohorts of breast cancer patients with a large panel of biomarkers to explore the clinicopathologic differences that exist between TNBCs that express one or more of basal markers and defined as BLBC (such as cytokeratin, CK, CK5/6, CK17, CK14, and EGFR, about 70%) and TNBCs that do not express these markers, being defined as TNBCKE- (about 30%) [141]. Although the morphologic features of BLBC and TNBCKE- were similar, these tumor subtypes differed for several clinicopathologic features: BLBCs are associated with the expression of hypoxia-associated factor (CA9), neuroendocrine markers, and markers, such as p53; BLBC tumors display more frequently BRCA1 alterations when compared to TNBCKE- (37% vs 4%); finally, the BLBC tumors show a unique pattern of tumor metastasis and respond better to chemotherapy, but they have a shorter survival when compared to TNBCKE- tumors [141]. Prat et al. estimated that 21% of TNBC are not basal-like and 31% of breast cancers showing basal-like gene expression are not triple-negative [142]. The basal-like subtype of breast cancer should be defined only on the basis of gene expression profiling assays and in the last WHO classification is recognized as one the intrinsic breast cancer subtypes [143]; according to the histological features, basal-like breast cancer is described as a tumor with a medullary pattern [144].
The analysis of the distribution of intrinsic subtypes among TNBCs according to PAM50 was basal-like (78-86%), luminal A (2-5%), luminal B (1-2%). HER2-enriched (6-15%) and normal-like (2-5%) [145].
Claudin-low is another intrinsic subtype that is associated with the TNBC phenotype. Claudin-low and metaplastic breast cancers, triple negative breast cancers, form a group enriched in tumors displaying EMT features. The molecular characterization of these tumors showed several interesting and peculiar findings: the loss of genes that are involved in cell-cell adhesion; enrichment for stem cell-like and EMT markers; and, frequent genomic aberrations that activate the PI3K/AKT pathways [146]. These peculiar molecular features represent an important support to understand the poor response to therapy and the cellular origin of this cancer subtype. Interestingly, the claudin-low and metaplastic breast cancer subtypes exhibit a gene expression signature, called “EMT core signature”, which is similar to that displayed by human mammary epithelial cells induced to undergo an EMT by expressing Snail, Twist or by TGF-beta1 [147]. Claudin-low are present in about 7–14% of all breast cancers [148]. Approximately 70% of claudin-low breast cancers are TNBC, with high frequency of metaplastic and medullary breast carcinomas; these tumors are characterized by low levels of cell adhesion molecules and elevated expression of immune-related genes, such as CD4 and CD79a, and by mesenchymal features (high expression of CD44, vimentin, and N-cadherin) and low epithelial differentiation (low CD24 expression), resembling a mammary stem-like phenotype, which was acquired by EMT [148]. Two recent studies have provided an extensive genomic, transcriptomic and clinico-pathologic characterization of claudin-low breast cancers, highlighting their consistent heterogeneity [149][150]. Both studies showed that claudin-low breast cancers are characterized by low genomic instability, mutational burden and proliferation levels and by high levels of immune and stromal cell infiltration [149][150]. A first study showed that claudin-low tumors are heterogeneous for their distribution at the level of intrinsic subtypes, the majority pertaining to basal-like and normal-like subtypes; about 15% of basal-like and normal-like breast cancers display phenotypic properties of claudin-low tumors, while about 1% or less of LumA, LumB and HER2-enriched breast cancers display a claudin-low phenotype [149]. The genetic alterations observed in claudin-low tumors largely reflect their correspondence to the various intrinsic subtypes [149]. A second study showed the consistent heterogeneity of claudin-low tumors that, according to genomic abnormalities and gene expression profile, can be subdivided into three subgroups: the subgroup 1 (CL1) is defined by a low (<10%) FGA (fraction of genome altered) and by the predominant presence of ER-negative tumors pertaining to the integrative cluster 4; the subgroup 2 (CL2) displayed an intermediate FGA level (from >10% to <30%) and were similar to luminal A tumors; the subgroup 3 (CL3) showed high FGA (>30%) and displayed features similar to those of basal-like tumors [150]. CL1 displayed an enrichment in stem cell-related signatures, CL2 in luminal-related signatures and CL3 in cell cycle-related pathways [150]. CL1 showed the highest enrichment in claudin-low related pathways [150]. The basal-like-related CL3 subgroup of claudin-low tumors was associated with low overall and disease-free survival [150].
Luminal epithelial cells can generate basal-like and claudin-low mammary tumors when endogenous or exogenous mutant RAS is expressed in an epithelial cell lineage-independent manner [151]. Continuous signaling of oncogenic RAS and regulators of EMT play a key role in the modulation of the plasticity and of the stem/mesenchymal properties of claudin-low breast cancers [151]. Neuropilin-1 (NP1) is highly expressed in claudin-low breast cancers and is required to maintain maximal RAS/MAPK signaling via EGFR and PDGFR; NRP1 inhibition in claudin-low tumors suppresses expression of stem and mesenchymal markers, such as ZEB1 and ITGA6 [152].
The histology of TNBCs is heterogeneous. The large majority (>85%) of TNBCs correspond to high-grade invasive carcinoma of no special type (NST) with special features of invasive breast cancers with invasive borders, a central necrotic area, lymphocytic infiltrates, and numerous mitoses [153]. In addition to these more frequent TNBC histological subtypes, rare histological types are observed, including carcinoma with medullary pattern (considered a subtype of basal-like subtype, associated with lymphocyte infiltration, a good prognosis), carcinoma with apocrine differentiation (characterized by increased androgen signaling), adenoid cystic carcinoma (characterized by the presence of a MYB-MF1B fusion gene and by a salivary gland phenotype with an indolent clinical behavior), secretory carcinoma (characterized by an ETV6-NTRK3 fusion gene and by a low to moderate histology with a favorable clinical course) and metaplastic carcinomas (characterized by metaplastic differentiation of neoplastic epithelial cells to squamous or mesenchymal cells, with a consistent degree of intra- and inter-tumor heterogeneity and consistent enrichment for genetic alterations of the Wnt and PIK3CA pathways; these tumors are preferentially classified as claudin-low tumors or basal-like tumors) [154][155].
Some recent studies were dedicated to the characterization of the molecular abnormalities occurring in 104 TNBCs at the time of diagnosis and they showed a consistent degree of heterogeneity, with some samples displaying coding somatic aberrations that were limited to few pathways, while other samples contained hundreds of coding somatic mutations [156]. The overall pattern of CNAs in TNBCs resembled that generally observed in breast cancers, with the most frequent CNA events occurring at the level of PARK2 (6%), RB1 (5%), PTEN (3%), and EGFR (5%) [156]. The analysis of gene mutations showed that p53 is the most frequently mutated gene (62% in basal TNBC and 43% in non-basal TNBCs); frequent mutations in PIK3CA (10.2%), USH2A (9.2%), MYO3A (9.2%), PTEN, and RB1 (7.7%) and ATR, UBR5, and COL6A3 genes (6.2%) are also observed [156]. The analysis of clonal distribution of mutations further supported the consistent heterogeneity of TNBCs: the basal subtype of TNBC displays some more variation than the non-basal TNBC; TP53, PIK3CA, and PTEN somatic mutations are clonally dominant over other genes; in other tumors, their clonal frequencies are incompatible with a founder status [156]. Finally, mutations in cytoskeletal, cell shape, and motility proteins occurred at lower clonal frequencies, thus implying that these genetic events occurred during tumor progression [156].
A study by Lehmann et al., which was based on an aggregate analysis of publically available expression data set, confirmed the consistent genetic heterogeneity of TNBCs, with the identification of seven subtypes: LAR-positive displaying a luminal pattern of gene expression (high expression of GATA3, FOXA1), with elevated androgen receptor (AR) levels, corresponding to luminal A and luminal B intrinsic subtypes; claudin-low-enriched mesenchymal, characterized by low claudin expression and enrichment in angiogenesis and stem cell-associated genes; mesenchymal stem-like, being characterized by expression profiles related to cell motility, differentiation, and EMT; immune response (IM), characterized by expression of genes that are involved in antigen presentation and processing, immune cell, and cytokine signaling; and two-cell cycle-disrupted basal subtypes: BL-1 (characterized by high expression of genes involved in DNA-damage response and cell-cycle regulation) and BL-2 (characterized by high levels of growth factor signaling and metabolic pathway activity); an unstable (UNS) cluster, being characterized by genetic instability [157]. The seven-subtype classification predicted a pathological response to neoadjuvant chemotherapy, but not overall survival in retrospective studies [158]. Another study of targeted ultra-deep sequencing that was carried out on 104 TNBCs confirmed the above reported findings, with clonal TP53 mutations being present in about 80% of samples and more subclonal mutations occurring at the level of the PI3K pathway (29.8%), MAPK signaling pathway (8.7%), and cell-cycle regulators (14.4%) [159].
A transcriptional study that was based on the analysis of 84 TNBC samples led to the identification of four TNBC subgroups that were associated with different clinical outcomes: luminal/AR, mesenchymal, basal-like/immune-suppressed, and basal-like/immune activated groups [160]. In this classification, a luminal androgen receptor (LAR) subtype is characterized by androgen receptor signaling; a basal-like subtype subtype is characterized by high immune cell signaling and cytokine signaling gene expression (BLIA); a basal-like and immune-suppressed (BLIS) subtype is characterized by an upregulation of cell cycle, the activation of DNA repair, and cytokine signaling gene expression; and, a mesenchymal-like (MES) subtype is characterized by enrichment in the expression of mammary stem cell pathways. In line with the previous study, TNBC tumors with tumors most expressing immune component features have the best outcome [160].
Subsequently, Lehmann et al. simplified their TNBC classification into seven groups and moved to a new classification in four groups: BL1, BL2, M, and LAR [161]. This change was justified by the observation that the histological analysis and laser microdissection before RNA isolation for gene expression studies provided evidence that the presence of stromal cells largely influenced the definition of the IM and MSL subtypes that were removed from the revised classification [161]. According to this classification, the four BL1, BL2, M and LAR subtypes differed in prognosis, response to chemotherapy, in initial patters of presentation and recurrence [161].
Jiang et al. have proposed a slightly different classification into four transcriptome-based subgroups: (i) a luminal androgen receptor (LAR) subtype (23%), being characterized by androgen receptor signaling; (ii) an immunomodulatory (IM) subtype (24%) with high immune cell signaling and cytokine signaling gene expression; (3) a basal-like and immune-suppressed (BLIS) (39%), characterized by the upregulation of cell cycle, activation of DNA repair, and downregulation of immune response genes; and, (iv) a mesenchymal-like (MES) subtype (15%) enriched in mammary stem cell pathways [162]. The LAR subtype was more frequent among Chinese TNBC patients compared to Caucasian TNBC patients [162]. Compared to the other TNBC subtypes, the LAR subtype displayed a lower frequency of TP53 alterations and a higher frequency of PIK3CA, KMT2C and PTEN gene alterations [162]. These authors also reported a very extensive characterization of molecular alterations that were observed in TNBC at the level of both somatic mutations and copy number alterations [162] (Figure 3). Ech avarria and coworkers have explored the pathological response of TNBC patients subdivided according to the simplified Lehmann’s classification to standard neoadjuvant chemotherapy regimen that is based on carboplatin and docetaxel and observed the highest response rates among BL1 (65.6%) tumors, followed by BL2 (47.4%), M (36.4%), and LAR (21.4%); the overall response rate was 44.7% [163].
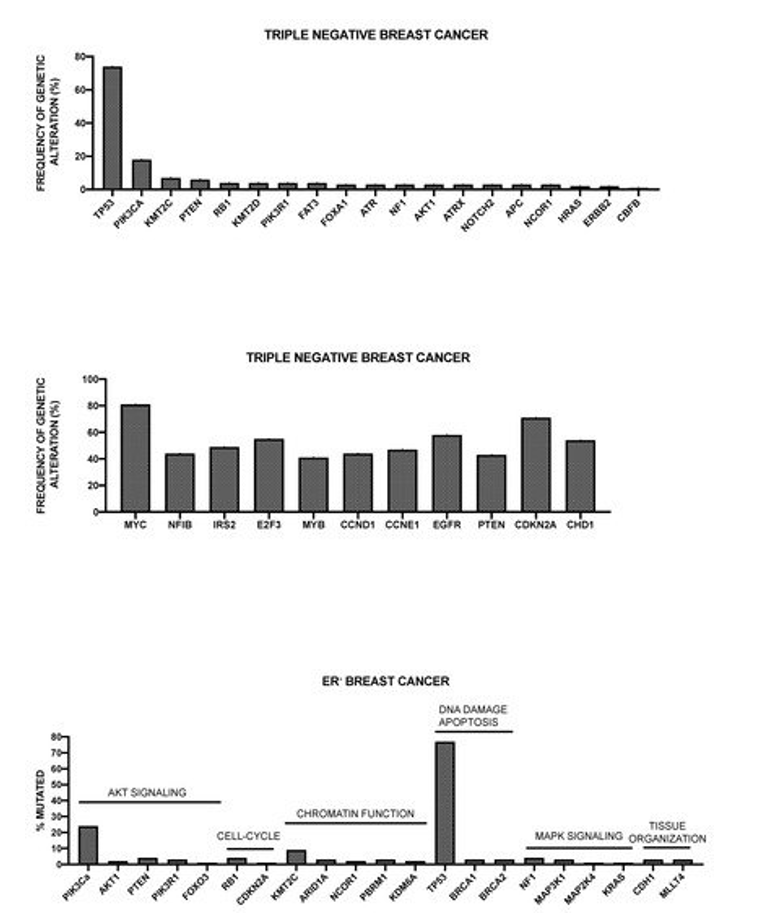
Figure 3. Genetic alterations observed in triple-negative breast cancers (TNBC). Top and middle panels: most recurrent somatic mutations (top panel) and copy number alterations (middle panel) observed in TNBC. The data shown are reported in Jiang et al. 2019 [162]. Bottom panel: most recurrent somatic mutations, subdivided according to functional gene groups, observed in estrogen receptor (ER)-negative breast cancers. The data shown are reported in Pereira et al. 2016 [51].
Bareche and coworkers analyzed the data that were related to TNBC patients on CNAs, somatic mutations, and gene expression contained in TCGA and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) and subdivided according to Lehmann’s classification, showing that: (i) BL1 subtype was identified as the most genomically unstable subtype (with the highest number of CNAs), with high TP53 mutation rates (92%) and copy number alterations in genes involved in DNA repair mechanisms (BRCA2, MDM2), AKT signaling (PTEN), and cell-cycle regulation (RB1) and high gain/amplifications of MYC, CDK6, and CCNE1; (ii) the LAR tumors were associated with higher mutational burden with enriched mutations at the level of the PI3KCA (55%), KMT2C (19%), AKT1 (13%), CDH1 (13%), NF1 (13%), and AKT1 (13%), and with higher frequency of gain/amplification of EGFR and AKT1; (iii) the M and MSL subtypes were associated with higher signature score for angiogenesis and low claudin expression; furthermore, the M subtype was associated with higher frequencies of gain/amplification levels of DNMT3A and TP53 and enriched for EGFR and NOTCH signaling; (iv) IM showed a high expression of immune signatures and check inhibitor genes, such as PD1, PDL1, and CTLA4 [164]. The presence of specific differences in mutational and CNA profiles between the various TNBC subtypes offers the way to potential therapeutic approaches [164]. Thus, the presence in the BL1 subtype of high genomic instability, high copy number losses for TP53, BRCA1/2, and RB1 genes, as well as copy number gains for PPAR1 gene, support the view that these tumors might be sensitive to PARP inhibitors. The pattern of genetic alterations of RB1 and of expression of CDK4 and CDK6 in LAR and MSL subtypes suggests a potential sensitivity to CDK4/6 inhibitors. EGFR and NOTCH signaling pathways are activated in the M subtype, suggesting a possible targeting of these signaling pathways in these tumors. Finally, the pattern of expression of immune checkpoint inhibitor genes in IM subtype suggests a possible benefit deriving from therapy with checkpoint inhibitors [164].
Other studies have identified genes, whose expression might help to classify TNBCs and predict their response to therapy. Thus, Quist and coworkers identified a four-gene decision tree signature, which robustly classified TNBCs into six subtypes; all four genes, EXO1, TP53BP2, FOXM1, and RSU1, in the signature are associated with either genomic instability, malignant growth and tumor progression, or treatment response [165]. One of these six subtypes, MC6, represented the largest part of tumors (about 50% of primary TNBCs); in metastatic TNBC patients, only 25% of the tumors pertain to the MC6 subtype and they are associated with a higher response rate to platinum-based chemotherapy [165]. Hsu et al., using a gene co-expression network analysis, identified the immunoglobulin-related genes IGHA1, IGHD, IGHG1, IGHG3, and IGLJ3 as the suppressor genes in the recurrence of TNBC patients; an immune score was established according to the expression of these six genes, being able to predict for the recurrence of TNBCs: as the score increases, the risk of recurrence decreases [166].
Jiang and coworkers performed a wide analysis of clinical, genomic, and transcriptomic data of a cohort of 465 of primary TNBC of Chinese patients [162]. These TNBCs have more frequent PIK3CA mutations and chromosome 22q11 copy-number gains than non-Asian TNBCs [162]. The LAR subtype showed more ERBB2 somatic mutations, infrequent mutational signature, and frequent CDKN2A loss [162]. LAR patients are candidates for combination therapy with the PI3K pathway and androgen inhibitors; furthermore, CDKN2A loss and CCND1 amplification may render LAR tumors potentially sensitive to CDK4/6 and AR inhibitors [162]. The molecular stratification of TNBC patients represented the background for the development of the TNBC umbrella clinical trial FUTURE, which combined the TNBC subtyping and genome-guided targeted therapy for refractory metastatic TNBC patients [167].
Using multi-omics datasets of TNBC, the heterogeneity of tumor microenvironment it was explored, providing evidence about three different clusters: cluster 1 was characterized by an incapability to attract immune cells, and MYC amplification was correlated with a low immune response; cluster 2 was characterized by chemotaxis, but inactivation of innate immunity and low tumor antigen burden, thus contributing to immune escape, in association with mutations of the PI3K-AKT pathway; cluster 3 was characterized by a high expression of immune checkpoint molecules [168]. These observations have some obvious potential therapeutic implications.
Tumors that were present in the public datasets, related to 494 TNBCs (153 treated with neoadjuvant chemotherapy) were evaluated for the development of a novel classification of these tumors. The tumors were subdivided into four subgroups, LAR, basal, claudin-low, and claudin-high, while using the cancer stem cell hypothesis as reference [169]. Samples with high luminal metanode activity were classified as LAR; tumor with low luminal metanode activity and high basal metanode activity were classified as basal; tumors low in both the luminal and basal metanodes were screened for claudin expression and then subdivided into a low-claudin and high-claudin subgroups, according to the claudin levels [169]. 18% of TNBC tumors corresponded to LAR, 11% to claudin-low, 63% to basal, and 8% to claudin-high subgroups [169]. This classification did not show any prognostic impact. Taking the immune metanode into account, the tumors were split according to their immune activity into low/high; in the whole TNBC population, 52% of tumors were IM-high and 48% IM-low: IM-low and IM-high tumors were present in all the four subgroups [169]. The immunological subdivision of LAR and claudin-high subgroups introduced a prognostic tool: in fact, LAR and claudin-high immune-negative TNBCs were associated with a significantly lower survival than the corresponding subgroups immune-positive; the immunological subdivision did not impact the prognosis of basal and claudin-low tumors [169].
Other studies were focused on defining the changes in genetic alterations that occurred in TNBCs. Tumor cells remaining after neoadjuvant chemotherapy contain cell populations that are intrinsically resistant to chemotherapy. Balko and coworkers have explored the molecular profiling of the residual disease of TNBCs after neoadjuvant chemotherapy and they have raised some observations: (i) when compared with basal-like primary tumors in the TCGA database, a higher frequency of MCL1 amplifications and a tendency to higher frequency of PTEN deletions or mutations and JAK2 amplifications in residual disease were observed; (ii) some of the molecular alterations in the residual disease after neoadjuvant chemotherapy, such as JAK2 amplification, and MYC amplifications predicted poor overall survival, while PTEN alterations were a favorable prognostic factor [170].
In another study, Jiang and coworkers have identified gene abnormalities conferring sensitivity to neoadjuvant chemotherapy in TNBC [171]. This study was based on the analysis of 29 TNBCs, in part (18/29) achieving a complete pathological response following neoadjuvant chemotherapy; the results of this analysis showed that, although mutations in single genes were not individually predictive, TNBCs exhibiting mutations in genes that are involved in AR and FOXA1 pathways were much more sensitive to chemotherapy [171].
A recent study reported the results of a comprehensive analysis of mutations, copy number, transcriptomic, epigenetic, proteomic and phosphor-proteomic patterns of TNBC subtypes [172]. This integrative analysis further supported the consistent heterogeneity of TNBC subtypes and showed the existence of specific pharmacologic vulnerabilities [172]. These studies suggested a different cellular origin of these subtypes: BL1 subtype was related to the bipotent L1.2 luminal progenitor cells; LAR subtype was related to the L2 hormone-responsive cells; the BL2 and M subtypes correlated to myoepithelial/basal cells [172]. The LAR tumor subtype displayed some peculiar properties, including a lower mutational burden, frequent PIK3CA and HER2 mutations, low levels of cell-cycle, DNA repair, MAPK, NOTCH and epigenetic gene alterations, increased protein and phospho-protein for AR, FOXA1, HER2, PIK3CA, and AKT; in line with these molecular features, LAR-subtype TNBCs displayed a low sensitivity to neoadjuvant chemotherapy [173]; in contrast these tumors exhibited clinical benefit deriving from treatment with the AR inhibitor enzalutamide and the PI3K inhibitor taselisib [174]. The BL2 subtype displayed frequent PI3K and MAPK mutations and rare DNA repair mutations, frequent PI3K/mTOR pathway activation, EGFR and MAPK signaling pathway activation (as supported by ERK1, ERK2 and MEK activation); BL2 tumors showed elevated CDK6 level [172]. Mesenchymal subtype displays frequent cell-cycle related gene mutations and DNA repair alterations, high mutation burden, genomic instability, absence of immune cells, low PDL1 expression, decreased global DNA methylation, and transcriptional repression of antigen presentation genes; in these tumors, major histocompatibility complex I (MHC-1) is transcriptionally suppressed by epigenetic mechanisms mediated by polycomb suppressor complex 2 (PRC2): pharmacological inhibition of PRC2 restores MHC-1 expression and enhances chemotherapy efficacy in murine tumor models, thus supporting the use of PRC2 inhibitors in the treatment of mesenchymal TNBCs [172]. LAR TNBC subtype is characterized by a tumor mutational burden, frequent PK3CA and HER2 mutations and rare mutations at the level of cell-cycle, DNA repair, MAPK, NOTCH and epigenetic regulators; these tumors are dependent on CDK4 and CCND1 [172].
PTEN loss is observed in about 20-25% of TNBCs. W.ang et al. have shown that PTEN loss and reduced expression of five miRNAs (miR-4324, miR-125b, miR-381, miR-145 and miR-136) identify a subgroup of breast cancers associated with very negative prognosis [175]. Interestingly, PTEN loss clusters almost exclusively with the TNBC BL1 subtype [176]. BL1 TNBC patients can be stratified into good-poor prognosis groups. Patients with PTEN-low within the BL1 group exhibit a particularly poor clinical outcome. This aggressive TNBC subgroup displays a unique signaling profile, characterized by high Rho-signaling [176]. RB1 loss and particularly high Rho A signaling identify a subset of BL1 patients with particularly poor outcome. Furthermore, in BL2 TNBC subtype, AKT1 copy number gain and high AKT1 mRNA levels were associated with poor clinical outcome [176]. Finally, in IM TNBCs, high PD1 mRNA levels were associated with poor outcome [176].
A proteomic analysis of archival breast cancer clinical specimens identified biological subtypes with different survival outcomes [177]. According to proteomic analysis four subgroups of TNBCs were defined: cluster-1 (basal-immune hot) is associated with the most favorable survival compared with other clusters and is characterized by enrichment in immune-response genes, antigen processing and presentation high TIL (tumor infiltrating lymphocytes) content; cluster-2 (mesenchymal) is associated with intermediate survival and was characterized by enrichment in ECM, blood coagulation and humoral immune-response genes; cluster-3 (luminal) was associated with intermediate survival and was characterized by enrichment for lipid metabolism, catabolic and oxidation-reduction processes and involves tumor specimens classified as HER2-enriched following PAM50 analysis; cluster-4 (basal-immune cold) displayed the poorest survival and was characterized by enrichment for DNA replication and cell-cycle protein genes and by the lowest T lymphocyte infiltrates [177].
Furthermore, mutations that lowered BRCA1 or BRCA2 RNA are associated with a better overall survival; a BRCA deficiency signature was defined and used to define a subset of chemosensitive TNBCs [178]. BRCA deficient tumors were characterized for a higher number of mutations per clone, a higher level of immune activation, and a higher mutational burden [178].
7. Breast Cancers with a Hereditary Component
A significant proportion of breast cancer patients have pathogenic germline mutations at the level of genes that increase the risk of developing a mammary neoplasia. Studies carried out in the last 10 years, based on large-scale sequencing studies have provided evidence about an association between germline protein-truncating variants and/or missense variants in 10 genes: ATM, BARD1, BRCA1, BRCA2, CDH1, CHEK2, PALB2, RAD51C, RAD51D, TP53 [179][180][181][182][183].
Several studies have shown the existence of a hereditary genetic component in a part of TNBC patients. Thus, over 80% of hereditary BRCA1-mutated breast cancers are classified as TNBC and about 15% of TNBCs occur in carriers of a BRCA germline mutation (gBRCA) [184][185]. The BRCA1 and BRCA2 gene products play an essential role in the activation and transcriptional regulation of DNA damage and cell-cycle control. Particularly, the BRCA1 and BRCA2 proteins exert a key role in DNA double-strand break repair by homologous recombination (HRR) and the maintenance of DNA stability. Breast cancer cells lacking functional BRCA1 or BRCA2 genes display a deficiency in HR repair of DNA double-strand breaks, determining a condition of dependence on alternative mechanisms to repair these DNA lesions and to genomic instability.
The results of a meta-analysis, including a large set of literature data, provided evidence that breast cancer patients with BRCA1Mut carriers were more likely to have TNBC than those of BRCA2Mut carriers or non-carriers [186]. Other genes and genetic elements, beyond BRCA1 and BRCA2, have been associated with an increased risk of TNBC [186].
A set of recent studies characterized the TNBC exhibiting deficiencies of homologous recombination repair (HRDs). HR is a biochemical pathway that coordinates the repair with the high-fidelity of double-stranded DNA breaks. HRD determines a condition of cellular dependency on alternative, error-prone DNA-repair pathways; the activation of this pathway determines the development of characteristic genomic alterations, higher mutational rates, and specific dependencies that can be exploited for therapeutic targeting of these tumors. Germline and acquired BRCA1 and BRCA2 alterations represent the most typical genetic alterations that determine a condition of HRD, leading to the inactivation of these tumor suppressors. Previous studies carried out in various tumors have shown that the loss of BRCA1 or BRCA2 determines a typical pattern of base-substitution mutations that are commonly called signature 3 [187]. In this context, Polak and coworkers have explored DNA signatures in a large cohort of 992 breast cancers and detected four recurrent signatures: (i) signature 1 (C > T at CpG sites); (ii) APOBEC-related signatures; (iii) signature 6, associated with microsatellite instability; and, (iv) a uniform signature, similar to signature 3, which is associated with BRCA1 and BRCA2 mutations [188]. In this study, 250/992 cases displayed a signature 3: 60 of these cases displayed 10 BRCA1, 29 BRCA2, 19 RAD51C, 2 PALB2 gene alterations that can be directly related to HRD [188]. The most frequent alterations involving BRCA1 imply BRCA1 germline biallelic mutations, somatic null mutations, epigenetic silencing, and mRNA downregulation; for the BRCA2 gene, the most frequent alterations involve germline biallelic mutations, gene deletion, and other undetermined germline alterations; RAD1C alterations are almost exclusively relatable to epigenetic silencing mechanisms [188]. Interestingly, a logistic regression model allowed for accurately detecting a sensitivity of about 99% BRCA1/BRCA2-deficient tumors, including both of those exhibiting the loss and functional deficiency of BRCA1/BRCA2 genes [189]. Wide-genome sequencing of matched germline/tumor DNA, coupled with the somatic mutational signatures, represent a very sensitive tool for the definition of the etiology of familial breast cancer and the prediction of HRD and consequent sensitivity to PARP inhibitors [190].
Staaf et al. used the HRD detect mutational-signature-based algorithm to screen a large population of TNBCs, allowing for the classification of these tumors into three different subgroups: HRD detect-high, HRD detect-intermediate and HRD detect-low [191]. Fifty-nine percent of these tumors were classified as HRD detect-high and 67% of them are explained by germline/somatic mutations of BRCA1/BRCA2, BRCA1 promoter hypermethylation, RAD51C hypermethylation, or biallelic loss of PALB2; this group displayed better outcome on adjuvant chemotherapy as compared to HRD detect-low patients [191]. HRD-detect intermediate patients are minority (about 5%) and they have the poorest outcomes [191]. Finally, HRD detect-low TNBCs display poor outcomes, frequent PIK3CA/AKT1 pathway abnormalities, and in 4.7% of cases were mismatch-repair-deficient [191].
Recent studies have defined the co-mutation profile and the clinico-pathological features of breast cancers associated with germline alterations of HR genes. Breast cancers associated with germline BRCA1 and BRCA2 mutations display some peculiar clinicopathologic features: (i) BRCA2-mutated tumors are frequently associated with generation of ER+, PR+, HER2- breast cancers, while BRCA1-mutated tumors are markedly associated with the generation of TNBCs; (ii) the frequency of both BRCA1 and BRCA2-mutated breast cancers is clearly higher in breast cancers with a family history and with early onset (diagnosed at an age of less than 40 years) compared to sporadic breast cancers [192]. Annunziato and coworkers reported the characterization of 80 BRCA1-mutated breast cancers and showed a co-mutation profile in these tumors characterized by frequent mutations of TP53 (65%) and PIK3CA (29%), MYC amplification (44%), together with several co-amplified genes, such as RAD21, EXT1, RECQL4, RSPO2, EPPK1, PLEC [188]. In addition to PIK3CA mutations, BRCA1-deficient TNBCs display heterozygous (36%) or homozygous (7.5%) PTEN loss; genetic alterations of MYC and PIK3CA/PTEN co-occur in 29% of all BRCA1-mutated TNBCs [193]. In experimental animal models, MYC and PTEN were shown to be potent drivers of BRCA1- associated cancerogenesis [193]. Single-cell carried out in mammary epithelial cells showed that these cells in carriers of pathogenic BRCA1 variants displayed an increased somatic mutational burden compared to age-matched mammary epithelial cells of controls with no genetically increased risk of breast cancer [194].
Through the binding to the BRCA1 and BRCA2 proteins, PALB2 protein facilitates homologous recombination repair for DNA double breaks. PALB2 germline mutations are observed in 0.6-2.7% in familial breast cancer and their presence considerably increases the risk of developing a breast cancer. Yang et al have explored 524 families bearing germline pathogenic variants of PALB2 gene and estimated a risk of developing a breast cancer to age of 80 years of 53% [195]. This observation showed the existence of a substantial association between germline PALB2 pathogenic variants and breast cancer. Li et al. have analyzed the spectrum of genetic alterations in 24 breast cancers associated with germline PALB2 mutations: these tumors were genetically heterogeneous: all these patients display a germline PALB2 mutation; 11/24 patients display PALB2 LOH; 5/24 patients, all without PALB2 LOH, show somatic PALB2 somatic mutations; the most frequent co-mutations were PIK3CA (29%) and TP53 (21%), mutually exclusive and not observed in patients displaying somatic PALB2 mutations [195]. Biallelic PALB2 alteration was observed in 16/24 cases and was associated with a HRD mutational signature; however, monoallelic PALB2 was not associated with a HRD signature [196]. These findings were confirmed by Ng et al. showing that biallelic inactivation of PALB2 is associated with a higher mutation load and a higher proportion of mutational signature 3 [197]. Patients with germline PALB2 alterations showed TP53 and PIK3CA co-mutation frequency of 11% and 22%, respectively [198]. Lee et al. analyzed 15 cases of individuals with germline PALB2 mutations developing invasive breast cancers: 10/15 displayed biallelic bi-allelic inactivation of PALB2 involving either loss of heterozygosity of the wild-type allele or somatic PALB2 mutation; 4/5 of the germline heterozygous PALB2 subjects displayed high HRD scores, suggesting that alternative mechanisms of PALB2 functional loss could be operating in these cases [198]. Hu et al. reported the molecular characterization of 480 familial cancer patients, including 143 familial breast cancer patients and 10 of these patients had mono-allelic germline PALB2 mutations: all these germline PALB2 mutations were high-risk stop-gain, frameshift, or splicing mutations; one of these ten patients had also somatic PALB2 mutations and two loss-of-heterozygosity of PALB2 [199]. These finding help to explain why some heterozygous PALB2 carriers have a HRD phenotype and develop a breast cancer. Checkpoint kinase 2 (CHEK2) is a serine-threonine kinase activated by double-strand DNA breaks and regulating cell-cycle. CHEK2 mutations are associated with an increased risk of developing breast cancer, with an absolute risk of up to 37% of developing a breast cancer by the age of 70 years [200]. Breast cancers developing in carriers of CHEK2 mutations are characterized by a high expression of hormone receptors, ER and PR [201]. Functional analysis of CHEK2 variants showed that while truncating variants cause CHEK2 dysfunction, only a part of missense mutations induces a significant CHEK2 dysfunction; the degree of CHEK2 kinase dysfunction deriving from genetic alterations correlates with an increased risk for breast cancer development [202]. Mandelker et al. reported the molecular characterization of 33 breast cancers developing in patients with germline CHEK2 mutations: 16 patients displayed high-risk variants and 16 low-risk (p. ile157thr) variants [200]. CHEK2-associated breast cancers are in large part hormone receptor positive. 81% of patients with high-risk CHEK2 germline variants exhibited LOH, compared to only 29% in those with low-risk CHEK2 germline variants [200]. CHEK2-associated breast cancers lacked a a dominant mutational signature 3, a genomic property typically observed in homologous recombination DNA repair deficiency [200]. A recent study explored the genomic status of 50 breast cancers associated with germline CHEK2 mutations: about 50% of these tumors display somatic loss of the wild-type CHEK2 allele; tumors without CHEK2 LOH are chromosomally stable, while tumors with LOH display a weak level of chromosomal instability, lower in comparison with BRCA1/2-associated breast cancers [203].
The kinase Ataxia-Teleangectasia (ATM) plays a key role in the mechanisms of DNA damage response and cell-cycle control. Heterozygous germline ATM mutations are observed in about 1% of the population and confer an increased breast cancer risk; particularly, the ATM missense variant c.7271T>G (p.V2424G) confers a high breast cancer risk, particularly for invasive ductal breast cancer [204]. At variance with the ATM V2424G variant, the ATM D1853V, L546V and S707P variants have the least predictive ability for increased breast cancer risk [205]. Breast cancers observed in individuals with germline ATM mutations display several peculiar clinicopathologic features: most (>60%) of AMT carriers displayed a positive breast cancer family history; breast cancer arising in carriers of deleterious ATM variants are not associated with large-scale genomic instability; about 26% of ATM germline breast cancers were bilateral; more than 50% of ATM germline breast cancers are luminal B-like/HER2-negative tumors [206][207]. Weigelt et al. reported the molecular characterization of 24 breast cancers in ATM germline carriers: 79% of these tumors displayed loss of heterozygosity of the ATM WT allele but none showed high activity of mutational signature 3, associated with defective HRD repair; furthermore, none of these tumors displayed TP53 somatic mutations [208].
Approximately 4% of breast cancers are diagnosed in women younger than 40 years old; these tumors usually exhibit unfavorable clinicopathologic features. Recent studies have explored the molecular characteristics occurring in very young women with breast cancer: Waks et al have characterized 92 breast cancers occurring in women <35 years old and observed that the frequency of pathogenic germline variants (the majority of cases displayed alterations in BRCA1 or BRCA2) was higher than in older women with breast cancer (23.9% vs 8.8%, respectively) [209]; Andrikopoulou et al reported the characterization of 32 young patients with breast cancer (<40 years old) and observed that germline pathogenic variants were more frequent in young women breast cancers compared to older breast cancers, particularly for that concerns CHEK2 (12% vs 3%, respectively) and BRCA1 (8% vs 3%, respectively) [210].
8. Breast Cancers with Homologous Recombination Deficiency
The identification of a condition of HRD in TNBC patients is important and clinically relevant because it is associated with a clinical response to platinum compounds or PARP inhibitors. Thus, a recent study showed that the HRD score predicts the response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer [211].
Echavarria and coworkers evaluated the response to neoadjuvant chemotherapy (NACT) with carboplatin and docetaxel in a cohort of TNBC patients, which were classified in subtypes according to Lehmann’s refined classification [163]. The response to NACT was significantly associated with Lehmann’s subtype, even in multivariate analysis with the highest pCR rate in BL1 (65%), followed by BL2 (47%), M (36%), and LAR (21%) [163]. The LAR subtype was predominantly composed by non-basal intrinsic subtype, while BL1, BL2, and M tumors mainly correspond to basal-like PAM50 intrinsic subtype [163].
These observations have represented the background for clinical studies attempting to verify the therapeutic activity of PARP inhibition in TNBC. Thus, the phase III Olympiad trial involved the enrollment of breast cancer patients with a metastatic disease and with a germline BRCA mutation and HER2 negativity and randomized to receive either Olaparib monotherapy or chemotherapy; median PFS was significantly longer in the Olaparib group than in the standard chemotherapy group (7.0 months vs 4.2 months, respectively, with a response rate of 59.9% vs 28.8%, respectively [212]. The subgroup of TNBC patients with germline BRCA mutant had an objective response rate of 55% and displayed a clinical benefit at the level of PFS in comparison with patients receiving the physician’s choice of treatment in the registration phase III trial of the PARP inhibitor olaparib (5.6 months vs 2.9 months, respectively) [212][213]. In the registration phase III trial of talazoparib tosylate, patients with germline BRCA mutation exhibited an objective response rate of 62% and a PFS of 5.8 months [214][215]. The sequencing studies carried out on tumor tissues from patients with BRCA1/BRCA2 mutations enrolled in the phase III EMBRACA trial showed that BRCA LOH status, DNA damage response gene mutational burden, tumor homologous recombination deficiency showed no association with talazoparib efficacy [216]. Monotherapy with PARP inhibitors had an efficacy that was limited to patients with BRCA mutations. A recent clinical trial evaluated the efficacy of a PARP inhibitor (niraparib) with an anti-programmed death receptor-1 in the treatment of advanced, metastatic TNBC [217]. A combination of niraparib with pembrolizumab (anti-PDL1) displayed anti-tumor activity in patients with advanced TNBC, particularly in patients with tumor BRCA mutations [217]. A recent study evaluated the efficacy of talazoparib in a neoadjuvant setting of 20 patients with operable breast cancer with germline BRCA-disease (15 patients were TNBC) [218]. The results of this study showed that neoadjuvant single-agent oral talazoparib once per day for six months without chemotherapy resulted in a high-rate (53%) of complete responses [218].
BRCA 1–2 mutations predispose TNBC not only to an enhanced sensitivity to PARP inhibitors, but also to platinum-containing agents. The TNT trial evaluated carboplatin in BRCA 1–2-mutated TNBC BRCAness subgroups [219]. In the unselected patient population carboplatin was not more active than docetaxel; in contrast, in patients with germline BRCA-breast cancers, carboplatin had double objective responses than docetaxel (68% vs 33%); such a therapeutic benefit was not observed with BRCA1 methylation, BRCA1 mRNA-low tumors, or a high score of a Myriad HRD assay [219]. De Talhouet et al. have explored the survival of breast cancer patients with germline BRCA1 or BRCA2 mutations and undergoing treatment with chemotherapy in a population of 925 breast cancer patients: 171 women carried a BRCA1 mutation, 95 a BRCA2 mutation and 659 were non-carriers; in the whole population of breast cancer patients a prolonged DFS was observed for BRCA carriers and a trend toward prolonged DSS [220]. In the TNBC group, BRCA carriers exhibited both prolonged DFS and DSS in response to chemotherapy treatment [220].
In the neoadjuvant setting, the addition of a PARP inhibitor to carboplatin and paclitaxel did not improve the proportion of TNBC patients achieving pCR [221]. A matched cohort study of this BrighTNess study showed that germline BRCA carriers did not have higher frequency of pathological responses than non-germline BRCA carriers when carboplatin ± veliparib was added to neoadjuvant chemotherapy [222]. Analysis of the long-term efficacy in BrighTNess study showed that addition of carboplatin to neoadjuvant chemotherapy significantly improved pathological response rate, with long-term event-free survival; the addition of veliparib did not improve the therapeutic response [223]. A recent trial (CALGB 40603) showed that the addition of carboplatin to neoadjuvant chemotherapy significantly increased the rate of pathological responses, but did not improve long-term overall survival [224].
The analysis of changes that are induced by neoadjuvant chemotherapy in TNBC cells was of fundamental importance to understand the mechanisms involved in chemoresistance. Thus, several studies have shown that genomic instability, as measured through the acquisition of point mutations only modestly changed in patients displaying chemoresistance following neoadjuvant chemotherapy, whereas dynamic changes at the level of the copy number alterations, such as focal amplifications, were frequently acquired during chemotherapy [225]. This observation is largely in line with the fundamental observation that the bulk of the genomic instability of TNBCs is, in large part, related to the somatic copy number alterations that are driven by TP53 loss and emerge as rapid, punctuated bursts during disease development [226]. This conclusion was based on the analysis of individual cells in 10 TNBC patients, providing evidence that, in each of these tumors, one to three major clonal subpopulations were identified, sharing a common evolutionary lineage; furthermore, a minor subpopulation of non-clonal cells was also identified and classified as metastable, pseudo-diploid or chormazemic [226]. Philogenetic and mathematical modeling suggested that the development of tumor heterogeneity was unlikely to be developed through the gradual accumulation of copy number alterations over time, but seemingly through punctuated events, occurring early during tumor evolution: thus, each tumor evolved from founder CNAs that were concurrently acquired in the early stages of tumor evolution and stably maintained during initial tumor development; no evidence of intermediate branching was observed when the tumors progressed from diploid to aneuploid genomes; some TNBC tumors showed evidence of divergent subclones, only occurring during later stages of tumor evolution and being only characterized by the acquisition of few (one to three) CNAs [226].
Kim and coworkers performed the analysis of chemoresistance evolution by single-cell deep-exome sequencing at DNA and RNA level; using this technique, 10 patients achieving pCR were studied and 10 patients in which malignant clones persisted after treatment [227]. This analysis allowed for reaching the fundamental conclusion that resistant genotypes were pre-existing and adaptively selected by neoadjuvant chemotherapy, while the transcriptional profiles were acquired by reprogramming in response to chemotherapy in TNBC patients [227]. Particularly, this fundamental study showed two patterns of genomic and phenotypic evolution following NAC, showing two different classes of clonal dynamics: extinction, involving the elimination of tumor cells by NAC, leaving only normal diploid cells after treatment; clonal persistence, being characterized by a large number of residual tumor cells with genotypes and phenotypes that were altered by chemotherapy [227]. The genotypic changes occurring following chemotherapy are related to CNAs that emerge after chemotherapy and pre-existing to treatment; these adapting copy number alterations are responsible for transcriptional reprogramming of relapsing TNBCs [227].
In line with these observations, Hancock et al. performed the analysis of 135 TNBC patients undergoing NAC and analyzed matched somatic genomes pre/post NAC of patients with residual disease post-chemotherapy [225]. Their main findings that were obtained through sequencing of tumors showed: (i) chaotic acquisition of copy gains and losses, including the amplification of prominent oncogenes (frequent events occurring in relapsing tumors were gains on 1q, 8q, and 10p with amplifications of MCL1, MYC, and GATA3 and loss of BRCA2, commonly in conjunction with FLT3 and RB1); (ii) absence of significant gains in deleterious mutations and insertion/deletions; (iii) gene transcriptome profiling analysis showed an enrichment of regulators of stem cell-like behavior and the depletion of immune signaling; and, (iv) analysis of the type of TP53 alterations present in tumors showed that TP53 loss (associated with low copy number), observed in 29% of cases, was associated with high oncogenic signaling and decreased overall survival: somatic gain in 18q were associated with poor prognosis, seemingly driving the upregulation of TGFβ signaling through SMAD2 [225].
9. Rare Histological Subtypes
Invasive Lobular Breast Carcinoma (ILC) ILC is the second most frequent histologic subtype of breast cancer, representing 10–15% of all breast cancers. ILC is a type of breast cancer that begins in the milk producing glands (lobules) of the breast. ILCs are typically ER and/or PR-positive. Several variants of ILC have been identified, including 103 classic ILC, solid ILC, alveolar ILC, tubule-lobular ILC, and pleomorphic ILC. Analysis of a large set of metastatic breast cancer patients showed that lobular histology is an independent negative prognostic factor [228]. Overall survival was better, worse or similar in patients with ILC with TNBC, Hormone receptor-positive/HER2-negative and HER2-positive metastatic breast cancer, respectively, compared with patients with IDC [228].
The common molecular feature of all ILC variants is the loss of the epithelial cell-cell adhesion molecule E-Cadherin encoded by CDH1 gene [229]. The dysregulation of cell-cell adhesion that is driven by lack of E-Cadherin protein is responsible for the histologic feature of ILC, consisting in small neoplastic disco-adhesive neoplastic cells invading the stroma in a single-file pattern. Genetic mutations causing a loss-of-function have been identified in all variants of ILC, with a frequency that ranges from 50% to 60% [230]. Mutations of the CDH1 gene are usually associated with the loss of 16q, the chromosome region where this gene is located [229]. The lack of E-cadherin protein expression is observed in approximately 90% of ILCs, confirming the highly non-cohesive morphological characteristics of this type of tumor. A recent study explored, in detail, the frequency of absent E-cadherin protein expression in tumors that can be classified as ILCs: about 4.5% of these tumors show E-cadherin expression, whereas 95.5% of these tumors display absent E-cadherin expression [231]. At variance, in breast cancers that were classified as invasive mammary with mixed ductal/lobular features, E-cadherin expression was absent in 40% of cases, whereas the remaining cases display E-cadherin expression with a heterogeneous pattern of positivity at the histological level [232]. Some rare breast cancers have CDH1 alterations and exhibit IDC morphology [231]. Recent studies that are based on next generation sequencing have elucidated the molecular abnormalities of ILCs. Ciriello and coworkers have analyzed 127 pure ILCs and 88 mixed ILCs/IDCs and then compared them to 490 ductal breast cancers (IDSc) and showed mutations targeting PTEN/FOXA1 and TBX3 as ILC-enriched features [233]. PTEN loss that is associated with increased AKT phosphorylation, which was highest in all ILC variants; interestingly, alterations acting upstream of AKT were identified in 40% of ILC cases and they were associated with increased AKT phosphorylation [233]. FOXA1, which is a key modulator of transcriptional activity of ER, controlling ER DNA binding through a modification of chromatin accessibility, is mutated in approximately 3% of IDCs and 7% of LICs [233]. Analysis of mRNA expression data allowed for classifying ILCs into three ILC subtypes, termed reactive-like, immune-related, and proliferative [233].
Desmedt et al. performed the genomic characterization of a large set of ILCs providing evidence that: (i) CDH1 was mutated in 65% of tumors; (ii) alterations in one of the key genes of PI3K pathway, PIK3CA, PTEN, and AKT1 were observed in more than 50% of cases; (iii) HER2 and HER3 were mutated in 5.1% and 3.6% of the tumors, respectively (these mutations activate EGFR/ERBB pathway); and, (iv) mutations in FOXA1 and ERS1 copy number gains were detected in 9% and 25% of cases, respectively [234]. All these alterations were more frequent in ILCs than in IDCs [234]. The histologic diversity of ILCs was associated with specific genetic alterations: (i) enrichment of HER2 mutations in the mixed, non-classic ILC variant; and (ii) ERS1 gains in the solid ILC variant [234]. The AKT1 mutations were associated with an increased risk of early relapse [234].
Some recent studies have explored the mutational profile of metastatic ILCs. The genes most frequently altered in metastatic ILCs largely overlapped with those reported in primary tumors; compared to primary ILCs, metastatic ILCs more frequently displayed TP53 (20% vs 9%), ERBB2 (19% vs 12%), ERS1 (15% vs 2%), FAT1 (9% vs 2%) and RFWD2 (8% vs 1%); furthermore, metastatic displayed a higher mutational burden, and more frequently dominant APOBEC mutational signatures than primary ILCs [235]. At variance with ERS1 alterations that are enriched both in metastatic ILCs and ILDs, NF1 alterations are enriched only in ILC metastases: NF1 alterations are frequently associated with loss-of-heterozygosity of the normal allele, are mutually exclusive with ESR1 mutations, are frequently polyclonal and are de novo acquired [236]. These observations suggest that NF1 alterations play role in endocrine therapy resistance [236].
Most of ILCs express receptors for both steroid hormones (estrogen and progesterone), while HER2 amplification or overexpression is rare in these tumors. A rare subset of ILCs does not express HR and HER2 receptors and is defined as TNBC lobular carcinoma: these tumors had aggressive phenotypes, with more pleomorphism [237]. At the molecular level, these tumors were characterized by: (i) a higher mutation frequency of CDH1 mutations than in TNBC-IDC (50% vs 11%, respectively); a lower frequency of TP53 mutations than in TNBC-IDC (15% vs 66%); (ii) a high frequency (53.8%) of alterations of the Estrogen-Related Receptor Alpha (ESRRA) gene [237]. Analysis of the intrinsic subtype classification showed that 42.9%, 39.3%, 7.1% and 10.7% of TNBC-ILCs can be classified as luminal A, HER2-enriched, normal breast-like and basal, respectively [237].
Pleomorphic Lobular Carcinomas (PLCs) account for about 15% of all ILCs and, at histological level, are characterized by a high degree of cellular pleomorphism, high mitotic index, eosinophilic cytoplasm, nuclear hyperchromasia and prominent nucleoli [238]. At genomic level, PLCs display very frequent alterations of CDH1 gene, frequent activating HER2 mutations (28%), HER2 amplification (21%), and PIK3CA mutations (49%); 94% of PLCs have alterations at the level of HER2, PI3K pathway and FGFR1 [238].
Studies on large cohorts of ILC patients have shown that 5-6% of these patients display germline pathological variants that predispose to breast cancer development [239]. This analysis showed that CDH1 and BRCA2 were associated with a high risk of ILC, CHEK2, ATM and PALB2 with moderate risk and BRCA1 and CHEK2 p.Ile157Thr were not associated with clinically relevant risk [239]. Compared with IDC, CDH1 germline mutations were more than 10-fold enriched, while BRCA1 germline mutations were reduced in ILC [239].
Metaplastic breast cancer (MpBC) is a rare and peculiar malignancy that accounts for approximately 0.2–5% of all breast cancers. The typical feature of MpBC is represented by the differentiation of the neoplastic mammary epithelium to a non-glandular component, usually squamous or mesenchymal. According to their differentiation properties, these tumors have been classified as squamous cell carcinoma, spindle cell carcinoma, mixed squamous and spindle, mesenchymal, or spindle cell and mesenchymal. Hormone receptor markers are negative in these tumors that can be classified as TNBCs. Gene expression profiling studies have shown that MpBCs pertain to the claudin-low subtype or to the basal-like subtype.
Recent studies have clarified the peculiarities of the genetic alterations that were observed in MpBCs. Initial studies of genomic analysis using array comparative genomic hybridization showed that MpBCs display greater genomic instability than other invasive breast cancers and they also show a high frequency of PIK3CA mutations, a finding suggesting that these tumors are different from other TNBCs, showing a low frequency of PI3KCA mutations [147]. Whole-exome sequencing studies provided evidence that MpBCs harbored frequent TP53 (69%) PIK3CA (29%), PIK3R1 (11%), ARID1A (11%), FAT1 (11%), and PTEN (11%) mutations; when compared with standard TNBCs, MpBCs significantly more frequently displayed mutations in PI3K/AKT/mTOR pathway (57% vs 22%) and canonical Wnt pathway (51% vs 28%) [240]. These findings were confirmed in several studies reporting the molecular alterations observed in MpBCs. Thus, in these studies, TP53 mutations were observed in 56–75% of cases, PIK3CA in 23–48% of cases, PTEN mutations in 11–25% of cases [241][242][243][244].
The frequency of TP53 mutations was lower in MpBC than in other TNBCs, while the frequency of PIK3CA, PIK3R1, PTEN, KMT2D, HRAS and ARID1A was higher in MpBC thsan in other TBCs [245]. The most frequent copy number variations observed in MpBC involve amplifications of MYC (17.3%), CCNE1 (5.9%), CCND1 (8.4%) and deletions of CDKN2A (19%), PTEN (14.9%) and RB1 (6.5%) [245].
TERT promoter mutations were observed in 15% of MpBC and are mutually exclusive with TP53 mutations and more frequently are associated with PIK3CA mutations compared to TERT-WT MpBCs [246].
Krings and Chen have performed a next generation sequencing study in 28 metaplastic carcinomas, which were subdivided into the main histologic subtypes subdivided into chondroid-matrix-producing carcinomas, spindle cell carcinomas, carcinomas with squamous, mixed spindle/squamous, and mixed metaplastic differentiation [247]. In the whole tumor cell population recurrent mutations were observed at the level of TP53 (64%), PIK3CA/PIK3R1 (61%), RAS/MAP kinase (25%), and TERT (25%) mutations [247]. A great mutational heterogeneity was observed in the various histological subtypes: (i) Chondroid-matrix producing carcinomas lacked PI3K and RAS/MAPK aberrations and TERT promoter mutations, when compared to 100%, 39%, and 39% of non-matrix-producing tumors, respectively; in contrast, TP53 mutations were observed in 90% of these tumors; (ii) TERT promoter mutations were particularly frequent (47%) in the spindle cell carcinomas and in tumors with squamous differentiation; (iii) PIK3CA mutations were particularly frequent in squamous and spindle/squamous tumors, whereas PIK3R1 mutations were particularly frequent (80%) in spindle carcinoma; and, (iv) spindle cell carcinomas lacked TP53 mutations [247]. Ng et al. obtained similar findings [240].
Interestingly, Dave and coworkers reported a very high frequency (97.5%) of mutations at the level of the ribosomal protein L39: the mutations of this protein increased the inducible nitric oxide synthase; this finding suggests a possible use of nitric oxide synthase inhibitors in these tumors [248].
Afkhami and coworkers recently explored the mutational and immune profiling of 21 MpBC patients and have evaluated possible correlations with survival: the most commonly altered genes were TP53 (68%), PIK3CA (42%), and PTEN (16%); for patients with PIK3CA mutations, relapse-free survival and overall survival were significantly worse than for those without; PD-L1 expression was associated with worse survival [249]. In line with these observations, Joneja et al. observed PD-L1 expression in a high proportion (46%) of metaplastic tumors [242].
McCart and Reed have performed the analysis of a large set of MpBC patients (347 patients) and have analyzed the mutation profile of a subset of these patients; this study showed that the most significant indicators of poor prognosis were large tumor size, loss of cytokeratin expression, EGFR overexpression and, for mixed MpBCs, the presence of more than three different morphologic entities within the tumor [250]. Exome sequencing studies confirmed the enrichment in these tumors of TP53 and PTEN mutations; intriguingly, these authors also observed concurrent mutations of TP53, PTEN, and PIK3CA [250].
Among the types of MpBCs, there is a subgroup of spindle cell tumors that resemble mesenchymal lesions, but exhibit an epithelial/myoepithelial immunophenotype; a molecular characterization of these tumors showed some peculiar features when compared to other metaplastic tumors: in 82% of cases a distinct chromosomal loss in the 9p21.3 region, including CDKN2A and CDKN2B was observed; the biallelic loss of the CDKN2A/CDKN2B region was observed in 50% of deleted cases; the expression of CDKN2A was absent in all cases with 9p21.3 loss; other genetic alterations frequently observed in other metaplastic breast cancer subtypes, such as TP53 mutations, were virtually absent in myoepithelial carcinomas, whereas PIK3CA mutations were present in 53% of myoepithelial breast cancers [251].
Few data are available on the mutational profiles of metastatic MpBCs. The analysis of a small cohort of metastatic MpBCs showed more frequent PIK3CA mutations and CCND1 and SOX9 alterations in metastatic MpBC than in primary MpBC [235]. Recently, Stradella and coworkers reported the genomic characterization of 21 primary-relapse paired samples of MpBC [252]. Mutations present in primary tumors are maintained in disease relapse/progression and only few new alterations emerged in the tumor samples of relapsing metaplastic breast cancers, such as MYC amplification [252].
10. Genetic Alterations in Metastatic Breast Cancer
Breast cancers can relapse, and their individual prognosis is strictly related to the timing after surgery and to the location (locoregional or distant) tumor relapses. According to the timing of relapse, breast cancer patients can be subdivided as early and late recurring BCs [81]. The incidence of late recurring breast cancers is is high in a subset of patients with ER+ disease [81]. The majority of cancer-related deaths in ER+ (83%) and in ER- (87%) occurred after the development of distant metastases. In ER+ breast cancers, higher tumor grade, number of positive lymph nodes and tumor size increased the risk of disease progression; in ER- disease, a longer time from surgery and occurrence of late relapse or distant relapse decreased the risk of breast cancer progression [81]. ER- patients displayed a higher risk of distant relapse and cancer death in the first five years after diagnosis; after this time, the risk decreased considerably. ER+ patients had a lower risk during the first five years, but longer in time. In ER- patients, the IntClust 4ER- displayed a persistent and increasing risk of relapse (0.3 risk at 5 years and 0.49 at 20 years); the IntClust 5 (HER2+ enriched) displayed a poor prognosis (0.48 risk at 5 years and 0.65 risk at 20 years); the IntClust 10 (basal-like-enriched) displayed a risk probability of relapse at 5 years of 0.33 and at 20 years of 0.37; therefore, the IntClust 4 and IntClust5 subgroups displayed a high risk of late recurrence [81]. The ER+ breast cancers are subdivided into two groups, one composed by patients with IntClust 3, IntClust 7, IntClust 8 and IntClust 4 associated with a better prognosis and another composed by patients with IntClust 1, IntClust 2, IntClust 6 and IntClust 9 associated with a poorer prognosis and a high risk of late recurrence [81]. The ER+ IntClusts associated with poor prognosis are characterized by frequent copy number variants: IntClust 1 (8.6% of ER+ tumors) displayed frequent amplification of chromosome 17q23 (focal amplification of RPS6KB1); IntClust 2 (4.5% of ER+ breast cancers) is characterized by amplification and overexpression of multiple oncogenes on chromosome 11q13, such as RSF1 amplification; IntClust 6 (5.5% of ER+ tumors) is characterized by focal amplification of ZNF703 and FGFR1 on chromosome 8p12; Int Clust (8% of ER+ tumors) was characterized by amplification of MYC at 8q24 [81]. ER- breast cancers displayed more frequent visceral metastases than ER+ tumors (brain/meningeal 27% vs 11%; pulmonary: 50% vs 41%), while bone metastases are more frequent m in ER+ tumors than in ER- tumors (71% vs 43%) [81].
Several studies have explored the spectrum of genetic alterations observed in metastatic breast cancer compared to primary breast cancer Angus and coworkers have recently reported a whole-sequencing study (WGS) on 625 patients with metastatic cancer, providing another fundamental contribution to the analysis of the molecular abnormalities related to the metastatic status of this tumor [49]. When compared to the WGS from 560 primary breast cancers [49], the median number of single nucleotide variants in metastatic cancers was significantly higher than in primary tumors; similarly, tumor mutational burden was higher in metastatic than in primary tumors [49]. The increase of TMB was related to both the progression to the metastatic condition and treatment exerting a pressure in tumor evolution [47]. The analysis of mutational signatures that are present in primary breast cancer contributes to the observed increased TMB in metastatic breast cancer; particularly, a shift from a more indolent age-related mutagenesis in primary cancers toward more APOBEC-driven processes in metastatic breast cancer was observed [49]. Using the ratio of non-synonymous to synonymous mutations, 21 potential driver genes were identified in metastatic breast cancers, including the key drivers TP53, PIK3CA, ESR1, GATA3, and KMT2C; as expected, TP53 was enriched in TNBC, whereas ESR1, PIK3CA, and GATA3 were more frequently mutated in ER+ metastatic breast cancers [49]. The comparison of the mutational frequency of these driver genes between primary and metastatic tumors showed that, in ER+ metastatic breast cancers, the mutational frequency of ESR1, TP53, NF1, AKT1, KMT2C, and PTEN genes was higher than in primary breast cancers [47]. Finally, this study allowed for defining a number of molecular events in metastatic breast cancers that could be targeted by specific treatments: 11% of patients displayed a high TMB that represents a biomarker to select patients for immunotherapy; 9% of patients exhibit a homologous recombination deficiency and they are thus candidate for poly-ADP ribose polymerase inhibitors and/or double-stranded DNA break-inducing chemotherapy; specific genomic alterations, for which the FDA-approved drugs are already available, were observed in 24% of patients [49].
Yates and coworkers have analyzed by whole genome sequencing 17 breast cancer patients displaying three different patterns of tumor evolution to bypass these limitations: synchronous axillary lymph node metastasis; distant metastasis; and, local relapse after treatment [253]. Locoregional relapsed tumors and distant metastases are usually associated with acquisition of additional driver mutations compared with the primary tumors, while synchronous lymph nodes metastases usually display the same driver mutations that were observed in primary tumors [253]. A careful characterization of metastatic breast cancers would be of fundamental importance for several reasons, including: (i) the identification of genomic drivers responsible for metastatic disease progression; (ii) the evaluation of the impact of tumor heterogeneity at the clinical level; (iii) the identification of the main biologic determinants responsible for variability of response to therapy at the level of individual patients; and, (iv) the discovery of new therapeutic targets [253]. Ravazi et al. have evaluated the genomic landscape of endocrine-resistant advanced breast cancers in a group of patients enriched in hormone receptor-positive tumors [254]. Particularly, in a group of patients exposed to hormonal therapy, an increased number in alterations of the mitogen-activated protein kinase (MAPK) pathway and in the ER transcriptional machinery was observed [254]. Particularly, 22% of post-hormonal therapy HR+HER2- breast cancers displayed non-overlapping alterations in one or multiple effectors of MAPK signaling or in MYC or other transcription factors; these mutations were mutually exclusive with hotspot mutations in ESR1 [167]. The MAPK alterations are involved in the mechanism of resistance to hormonal therapy [254]. Activating ERBB2 mutations and NF1 loss-of-function mutations were more than twice more common in endocrine-resistant than in endocrine-responsive tumors [254]. Importantly, the global evaluation of endocrine-resistant breast cancers suggests a taxonomy categorizing these tumors into four groups: (i) tumors bearing ESR1 mutations; (ii) tumors with molecular alterations at the level of the MAPK pathway; (iii) tumors with genetic alterations at the level of the molecular machinery involved in transcriptional regulation; and (iv) pan-wild type tumors with an unknown mechanism of resistance [254]. Bertucci and coworkers have recently reported a detailed analysis of the genomic landscape of 617 metastatic breast cancers, being mainly represented by HR+/HER2- (381) and TNBC (182) [255]. The most frequently altered genes were TP53 (47%), PIK3CA (30%), GATA3 (18%), ESR1 (17%), KMT2C (10%), CDH1 (9%), PTEN (7%), and NF1 (7%); in the HR+/HER2- breast cancers, nine genes were the most frequently mutated in the metastatic setting, including TP53 (29%), ESR1 (22%), GATA3 (18%), KMT2C (12%), NCOR1 (8%), AKT1 (7%), NF1 (7%), RIC8A (4%), and RB1 (4%), with the TP53 and ESR1 genes being mutually exclusive [168]. TP53, NF1, and RB1 mutations were associated with poor outcome in HR+/HER2- patients [255]. HR+/HER2- metastatic breast cancers also displayed an increase in mutational signatures S2, S3, S10, S13, and S17, some of which are associated with poor outcome [255]. Importantly, the metastatic tumors show a significant increase in mutational burden and clonal diversity when compared to primary breast cancers [255]. In a precedent study based on the analysis of a smaller number of metastatic breast cancer patients, Lefebvre and coworkers identified twelve genes (TP53, PIK3CA, GATA3, ESR1, MAP3K1, CDH1, AKT1, MAP2K4, RB1, PTEN, CBFB, and CDKN2A) as significantly more mutated in metastatic breast cancer than in primary tumors [256].
Paul et al. have performed whole exome and shallow whole exome sequencing in metastatic breast cancer and in corresponding primary tumors. Through this approach, they identified seven genes that were preferentially mutated in metastases, ESR1, MYLK, PEAK1, SLC2A4RG, EVC2 and PALB2; four regions were preferentially copy-number altered in metastatic tumors, including loss of STK11 and CHKN2A/B and gains of PTK6 and the membrane-bound progesterone receptor, PAQR8 [257]. Interestingly, PAQR8 gains are mutually exclusive with ESR1A mutations, supporting their potential role in resistance to endocrine therapy [257]. Some pathways are preferentially altered or mutated in metastatic breast cancer, including cAMP/PKA, WNT, mTOR, CDK/RB, focal adhesion [257].
ESR1A mutations are one of the genetic alterations more linked to the development of breast metastatic status. In metastatic ER+ breast cancer, ERS1 mutations are observed in 20-30% of patients; Y537S and D538G are the most frequent ESR1 mutations. In addition, there are also some rare missense hotspot ESR1 mutations with lower phenotypes compared to Y537S and D538G mutations. These mutations result in ligand-independent constitutively activated ER, leading to increased proliferation and decreased sensitivity to endocrine treatments. These mutations are rarely observed in primary breast cancers (0-3%) but are more frequently observed in metastatic tumors: a higher frequency of ESR1 has been observed among metastatic patients treated with various lines endocrine therapy (20-30%) compared to early metastatic patients (6-10%) [258]. A high frequency of ESR1 mutations were observed in patients treated with endocrine therapy, in newly metastatic patients (12%) and particularly in those with advanced metastases (18%): the majority of these patients received tamoxifen or tamoxifen and aromatase inhibitors [259]. 36% of ESR1 mutations were observed among patients with local recurrence, but only 10% of these mutations were detected at allele frequency >1% [259]. The most frequently occurring ERS1 mutations, Y537S and D538G, confer peculiar regulating properties to the mutant Erα: in fact, these mutants, in addition to regulate a similar set of genes such as WT Erα, also displayed peculiar transcriptional activities resulting in the expression of genes encoding proteins promoting an aggressive tumoral phenotype associated with metastasis induction [260].
Li and coworkers have explored ESR1 mutations in four cohorts of metastatic breast cancer patients and observed the preferential occurrence of these mutations in patients with distal metastases [261]. ESR1 mutant breast cancers displayed a unique profile of transcriptome-associated with multiple metastatic pathways and, particularly with strong expression of cell-cell adhesion-related genes [261]. The reprogrammed cell adhesive gene network through alterations in desmosome/gap junction genes and the TIMP3/MMP axis, whose activation induces enhanced cell-cell contact and concomitantly decreased cell-extracellular matrix adhesion; these changes were reflected at clinical level by increased clusters of circulating tumor cells enriched in ESR1-mutated breast cancer cells [261]. Studies in suitable experimental models supported a cooperative therapeutic effect of co-targeting of Wnt and ER pathways [261].
ESR1 mutations were observed in 9% of metastatic ILC breast cancer patients; 6% of patients displayed ESR1 mutations in primary or axillary metastases, not detected in the matched distant metastases [262].
The results of two recent clinical trials directly support a role for ESR1 mutations in driving breast cancer evolution and resistance to endocrine therapy. The PADA-1 phase III trial evaluated the role of the presence of ESR1 mutations in the treatment of metastatic ER+ breast cancer: the presence of ESR1 mutations significantly decreased the median progression-free survival in first-line treatment with an aromatase inhibitor and the CDK4/6 inhibitor, Palbociclib (with a median progression-free survival of 11 months in the patients with ESR1 mutations and of 26.7 months in the group ESR1-WT [263]. After this initial step of treatment, the patients were evaluated every two months for the presence of ESR1 mutations by liquid biopsy analysis: rising ESR1 mutant patients were included in the second step, in which they were randomized to receive the continuation of standard therapy (standard arm) or switching to fulvestrant-palbociclib (experimental arm); the median progression-free survival was 5.7 months in the standard arm compared to 11.9 months in the experimental arm [264]. These results support the clinical utility of monitoring ESR1 mutations. In the phase III, randomized, double-blind PALOMA-3 study, patients who progressed on an aromatase inhibitor were randomized to Palbociclib plus fulvestrant versus fulvestrant alone: plabociclib plus fulvestrant demonstrated significantly improved efficacy compared to placebo plus fulvestrant, with a median overall survival of 11.2 months versus 4.6 months, respectively [265]. Patients in both arms of treatment showed an increase in the frequency of Y537S mutations; baseline ESR1 mutations were lower among long-term responders in both arms [265]. The 5-year overall survival rate was 23.3% with plabociclib plus fulvestrant and 16.8% with fulvestrant alone [266].
Several recent studies have explored the genetic alterations in metastatic breast cancers according to the body location of metastases. Metastatic breast cancer cells preferentially metastasize to specific metastatic sites through a process known as organotropism, with a metastatic site distribution involving multiple mechanisms mainly related to the subtypes of breast cancer cells, the molecular abnormalities present in these tumor cells and the interaction between tumor cells and tissue microenvironments [267]. The main tissue locations of breast cancer cells at the level of the bone (47-60%), liver (19-20%), lung (16-34%) and brain (10-16%) [267]. Molecular subtypes of breast cancer display differences in metastatic patterns; HR-/HER2+ ductal breast cancer had a higher prevalence of brain metastases than the HR+/HER2- subtype (38% vs 20%), while the latter had a higher prevalence of bone metastases (67% vs 49%) [268].
Chen et al. have reported a comprehensive characterization of metastatic breast cancers through the integration of the data of molecular characterization of primary and metastatic breast cancers from eight different cohorts [269]. The pan-metastasis analysis showed that ten of the most frequently altered genes in metastatic breast cancers did not display significant differences in their frequency compared to primary tumors, except for ESR1, ARID1A and NF1 which were more frequently altered in metastatic than in primary tumors [268]. 11 genes (ESR1, SMARCA4, NF1, FGFR4, ARID2, PARP1, TSC2, ATRX, ARID1A, AURKA, STAG2) were frequently identified in metastatic breast cancers; ARID2 mutations were more frequent both in treatment-naïve and post-treatment metastatic breast cancer samples compared to primary cancers, while ERS1 and NF1 mutations were significantly frequent only in post-treatment metastatic breast cancer samples [269]. Metastatic site-specific analysis showed that four altered genes, ESR1, CDH1, RICTOR, TP53 displayed a preference for at least one metastatic site: ESR1 and TP53 showed preference for metastatic sites in opposite direction (in the liver metastases, ESR1 mutations are frequent and TP53 mutations are rare, while an opposite trend was observed in lymph node metastases); RICTOR mutations are preferentially observed in bone metastases; CDH1 mutations are preferentially observed in ovarian and peritoneal metastases [269].
Rinaldi and coworkers reported the analysis of the genomic landscape of 11,616 breast cancers, including 5,034 metastases, undergoing targeted gene sequencing during standard clinical care [269]. In addition to the findings observed also in other studies on the molecular characterization of metastatic breast cancers, these authors reported, in addition to the two frequent hot spot mutations, a consistent metastatic enrichment of lower-frequency mutations of the ESR1 gene located at the level of the ligand binding domain [270]. The ESR1 mutation rate was highest in liver metastases (44%), followed by pleura and lung (24-25%) and bone (20%); visceral mutations have more D538G mutations than other ESR1 hotspot mutations; bone metastases displayed more Y537S than D538G mutations [270]. Some genetic alterations resulted enriched at specific metastatic sites: ASXL1 amplifications and PTEN deletions are enriched in brain metastases; DNMT3A mutations are enriched in bone metastases compared to other metastatic sites; NOTCH1 mutations are enriched in skin metastases; KRAS, KEAP1, STK11 and EGFR mutations are enriched in lung metastases [270].
Jiang et al. have developed a machine learning computational framework MetaNet, integrating clinical and sequencing data from 32,176 primary and metastatic cancer cases, to assess and predict the risk of metastatic progression of primary tumors [271]. MetaNet provided a high accuracy in distinguishing the metastasis from the primary in breast cancers: from this predictive system, Metastasis-Featuring Primary (FMP) tumors were identified as a subset of tumors with genomic features enriched in metastasis and with a higher metastatic risk and a shorter overall survival [271]. Some genomic alterations were associated with organ-specific metastases: ESR1 mutations is the top liver-tropic variant, while CDH1 mutation is the bone-tropic variant and TP53 mutation, CDK12 and HER2 amplifications are the brain-tropic variants [271].
Hu et al. have reported the results of a detailed whole-exome sequencing analysis involving paired primary tumors and metastatic samples from breast cancer, colorectal and lung cancer patients [272]. Breast cancers exhibited higher prevalence of both monoclonal-private clonal and subclonal driver mutations as compared to colorectal and lung cancers: 59% of clonal drivers were shared between primary and metastatic breast cancers, with 23% of subclonal drivers [272]. 53% of treated breast cancer metastases harbored one or more private clonal driver mutations, compared to only 22% in untreated metastases [272]. Copy number alterations were largely concordant between primary and metastatic tumors, with the exception of amplifications of IL7R, NIPBL and deletions of NOTCH1 and PTEN [272]. Polyclonal seeding was relatively common to untreated lymph node metastases and distant metastases, but less frequent in treated distant metastases [272]. These observations suggest that treatment promotes clonal evolution.
Nguyen et al. reported the results of a large genomic profiling and clinical information on metastatic events and outcomes of >25,000 cancer patients profiled at Memorial Sloan Kettering using the MSK-IMPACT targeted sequencing platform which identifies somatic mutations, rearrangements, copy-number alterations in 468 cancer genes, as well as tumor mutational burden (TMB), chromosomal instability and microsatellite instability [273]. High TMB seems to be a driver of metastases in both ductal and lobular breast cancers: the percentage of patients with a high TMB (≥10 mut/Mb) in metastases was higher both in HR+/HER2- breast cancer (2% vs 7%) and lobular breast cancer (5% vs 19%) [273]. In HR+/HER2- breast cancer ESR1 and TP53 mutations are positively correlated with metastatic burden, while in these tumors CBFB mutations are inversely correlated with metastatic burden [273]. In breast lobular carcer, ESR1 mutations are positively and CDH1 mutations are inversely correlated with tumor metastatic burden [273]. In breast ductal HR-/HER2+ cancer, CCND1/FGF19 and cell cycle alterations are positively correlated with tumor metastatic burden [273].
Liver metastases related to metastatic breast cancer have the second worst outcome after brain metastases; up to 50% of breast cancer patients have the probability to develop liver metastases during evolution of their tumoral disease. Tian et al have explored the genomic landscape of liver metastases in a cohort of Chinese patients and in a cohort of 217 breast cancer patients with liver metastases from Memorial Sloan Kettering Cancer Center [270]. Compared with the mutated genes observed in primary breast cancers, 14 genes, including ESR1, ARID2, BLM, FGFR4, APC, HER2, ROS1, ATR, IGF1R, NF1, JAK1, FAT1, NOTCH2 and AKT, display mutation frequencies significantly different in liver metastases compared to their corresponding primary tumors [270]. TP53 (43% of positivity) and ESR1 (20% of positivity) mutations in liver metastases are mutually exclusive [274].
Several studies have explored the changes occurring in breast cancer cells following their brain metastatic spreading. Breast cancer cells have the capacity to adapt to microenvironmental changes or to the therapeutic pressure. Several studies have shown that brain metastases diverge from primary tumors at phenotypic and genotypic level [275]. An initial study by Priedigkeit et al. explored the molecular features of brain metastases isolated from 20 metastatic breast cancer patients: 17/20 brain metastases retained the PAM50 subtype of the primary tumors; 17/20 brain metastases displayed significant changes in gene expression profile compared to primary tumors, including gains of FGFR4 (30% of cases), AURKA (10%), loss of ESR1 expression (45%); furthermore, about 20% of metastases acquired HER2 expression compared to their correspondent primary tumors [275]. A transcriptomic characterization showed that 71% of brain breast cancer metastases display elevated HER2 signature compared to the matched primary tumors [276]. Tumor switching from HER2-negatice to HER2-positive status displayed intermediate HER2 scores in the primary tumors; furthermore, loss of ESR1 expression correlated with increases in HER2 signature [276]. The most recurrent expression gains in brain metastases occurred at the level of RET and HER2 genes (both in 38% of cases) [276]. The analysis of 219 patients with brain metastases showed a global receptor (ER, PR, HER2) discordance of 36%; baseline subtype switching occurred in 37% of HR-positive patients and 12% of HER2-negative patients in primary tumors acquired HER2 mutations in brain metastases [277]. Loss of receptor expression in brain metastases was associated with worse prognosis [277].
A recent study reported the results of an extensive molecular characterization of 45 brain breast cancer metastases and of their matched primary tumors [278]. According to the transcriptomic and genomic profile, breast cancer tissues were classified into 11 different mutational signatures (from A to K); three signatures were significantly enriched in brain metastases compared to their matched primary tumors: Breast A (mismatch repair deficiency); Breast F (with associated driver mutations TP53, APC, NOTCH and NFE2L2); Breast K (HRD-related and associated with BRCA2, TP53, BRCA1, MYC, ARID1, NF1 driver mutations) [278]. The HRD mutational signature was independent of known germline and somatic BRCA1/2 and PALB2 mutations [278]. Experiments with PARP inhibitors in patient-derived brain metastatic tumor explants supported the functional relevance of this pathway and its vulnerability [278].
This entry is adapted from the peer-reviewed paper 10.3390/medsci8010018
References
- Forouzanfar, M.H.; Foreman, K.J.; Delossantos, A.M.; Lozano, R.; Lopez, A.D.; Murray, C.J.L.; Naghavi, M. Breast and cervical cancer in 187 countries between 1980 and 2010: A systematic analysis. Lancet2011, 9801, 1461–1484.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics-2019. CA Cancer J. Clin.2018, 68, 7–30.
- Breast Cancer Facts and Figures 2017–2018; American Cancer Society: Atlanta, GA, USA, 2017.
- Petersen, O.W.; Polyak, K. Stem cells in human breast. Cold Spring Harb. Perspect. Biol.2010, 2, a3160.
- Spike, B.T.; Engle, D.D.; Liu, J.C.; La, J.; Wahl, G.M. A mammary stem cell population identified and characterized in late embryogenesis reveals similarities to human breast cancer. Cell Stem Cell2012, 10, 183–197.
- Shehata, M.; Teschendorff, A.; Sharp, G.; Novcic, N.; Russell, I.A.; Avril, S.; Prater, M.; Eirew, P.; Caldas, C.; Watson, C.J.; et al. Phenotypic and functional characterization of the luminal cell hierarchy of the mammary gland. Breast Cancer Res.2012, 14, R134.
- Van Keymeulen, A.; Fioramonti, M.; Cantonze, A.; Bouvencourt, G.; Schouri, Y.; Blanpain, C. Lineage-restricted mammary stem cells sustain the development, Homeostasis, and regeneration of the estrogen receptor positive lineage. Cell Rep.2017, 20, 1526–1532.
- Wang, C.; Christin, J.R.; Oktay, M.H.; Guo, W. Lineage-biased stem cells maintain estrogen-receptor-positive and-negative mouse mammary luminal lineages. Cell Rep.2017, 18, 2825–2835.
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature2011, 479, 189–193.
- Zang, Y.A.; Nusse, R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell2010, 6, 568–577.
- Amerongen, R.; Bowman, A.; Nusse, R. Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell2012, 11, 387–400.
- Wang, D.; Cai, C.; Dong, X.; Yu, Q.C.; Zhang, X.O.; Yang, L.; Zeng, Y.A. Identification of multipotent mammary stem cells by protein C receptor expression. Nature2015, 517, 81–84.
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature2014, 506, 322–327.
- Cai, S.; Kalisky, T.; Sahoo, D.; Dalerba, P.; Feng, W.; Lin, Y.; Qian, D.; Kong, A.; Yu, J.; Wang, F.; et al. A quiescent Bcl11b high stem cell population is required for maintenance of the mammary gland. Cell Stem Cell2017, 20, 147–260.
- Trejo, C.L.; Luna, G.; Dravis, C.; Spike, B.T.; Wahl, G.M. Lgr5 is a marker for fetal mammary stem cells, but is not essential for stem cell activity or tumorigenesis. NPJ Breast Cancer2017, 3, 16.
- Fu, N.Y.; Nolan, E.; Lindeman, G.J.; Visvader, J.E. Stem cells and the differentiation hierarchy in mammary gland development. Physiol Rev 2020, 100, 489-523.
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggings, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic non stem cells can spontaneously convert to a stem like state. Natl. Acad. Sci. USA2011, 108, 7950–7955.
- Assalin-Labat, M.L.; Vaillant, F.; Sheridan, M.J.; Pal, B.; Wu, D.; Simpson, E.R.; Yasuda, H.; Smyth, G.K.; Martin, T.J.; Lindeman, G.J.; et al. Control of mammary stem cell function by steroid hormone signaling. Nature2010, 465, 798–802.
- Joshi, P.A.; Jackson, H.W.; Beristain, A.; Grappa, M.; Mote, P.; Clarke, C.; Stingl, J.; Waterhouse, P.; Khokha, R. Progesterone induces adult mammary stem cell expansion. Nature2010, 465, 803–808.
- Schmarek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.; Lee, H.; Hanada, R.; Joshi, P.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANK L controls development of progestin-driven mammary cancer. Nature2010, 468, 98–102.
- Nolan, E.; Vaillant, F.; Branstetter, D.; Pal, B.; Giner, G.; Whitehead, L. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med 2016, 22, 933-939.
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell2012, 148, 1015–1028.
- Balko, J.M.; Miller, T.W.; Morrison, M.M.; Hutchinson, K.; Young, C.; Rinehart, C.; Sanchez, V.; Jee, D.; Polyak, K.; Prat, A.; et al. The receptor tyrosine kinase ErbB3 maintains the balance between luminal and basal breast epithelium. Natl. Acad. Sci. USA2012, 109, 221–226.
- Kuperwasser, C.; Chevarria, T.; Wu, M.; Magrane, G.; Gray, J.W.; Carey, L.; Richardson, A.; Weinberg, R.A. Reconstruction of functionally normal human and malignant human breast tissues in mice. Natl. Acad. Sci. USA2004, 101, 4966–4971.
- Proia, D.A.; Kuperwasser, C. Reconstruction of the human mammary tissues in a mouse model. Protoc.2006, 1, 206–214.
- Eirew, P.; Stingl, J.; Raouf, A.; Turashvili, G.; Aparicio, S.; Emerman, J.T.; Eaves, C.J. A method for quantifying normal mammary human epithelial stem cells with in vivo regenerative ability. Med.2008, 14, 1384–1389.
- Giraddi, R.R.; Chung, C.Y.; Heinz, R.E.; Balcioglu, O.; Trejo, C.L.; Dravis, C.; Hagos, B.M.; Mehrabad, E.M.; Rodewald, L.W.; Hwang, J.Y.; et al. Single-cell transcriptomes distinguish stem cell state changes and lineage specification programs in early mammary gland development. Cell Rep.2018, 24, 1653–1666.
- Nguyen, Q.H.; Pervolarakis, N.; Blake, K.; Ma, D.; Davis, R.T.; James, N.; Phung, A.T.; Willey, E.; Kumar, R.; Jabert, E.; et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Commun.2018, 9, 2028.
- Wuidart, A.; Sifrim, A.; Fioramonti, M.; Matsumura, S.; Brisebarre, A.; Brown, D.; Centonze, A.; Dannau, A.; Dubois, C.; Van Keymeulen, A.; et al. Early lineage segregation of multipotent embryonic mammary gland progenitors. Cell Biol.2018, 20, 666–676.
- Liljia, A.M.; Rodilia, V.; Huyghe, M.; Hannezo, E.; Landgrain, C.; Renaud, O.; Leroy, O.; Rulands, S.; Simons, B.D.; Fre, S. Clonal analysis of Notch1-expressing cells reveals the existence of unipotent stem cells that retain long-term plasticity in the embryonic mammary gland. Cell Biol.2018, 20, 677–687.
- Chen, W.; Morabito, S.J.; Kessenbrock, K.; Enver, T.; Meyer, K.B.; Teschendorff, A.E. Single-cell landscape in mammary epithelium reveals bipotent-like cells associated with breast cancer risk and outcome. Biol.2019, 2, 306.
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. Clin. Oncol.2010, 28, 3271–3277.
- Wirapati, P.; Sotiriou, C.; Kunkel, S.; Farmer, P.; Pradervand, S.; Haibe-Kains, B.; Desmedt, C.; Igniatidis, M.; Sengstag, T.; Schutz, F.; et al. Meta-analysis of gene expression profiles in breast cancer: Toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res.2008, 10, R65.
- Lindström, L.S.; Karlsson, E.; Wilking, U.; Johansson, U.; Hartman, J.; Lidbrink, E.; Hatschek, T.; Skoog, L.; Bergh, J. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and epidermal growth factor receptor 2 are unstable throughout tumor progression. Clin. Oncol.2012, 30, 2601–2608.
- Early Breast Cancer Trialist Collaborative Group. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomized trials. Lancet2011, 378, 771–784.
- Early Breast Cancer Trialist Collaborative Group. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: Meta-analysis of individual patient data for 10,801 women in 17 randomized trials. Lancet2011, 3778, 1707–1716.
- Early Breast Cancer Trialist Collaborative Group. Comparison between different polychemotherapy regimens for early breast cancer: Meta-analyses of long-term outcome among 100,000 women in 123 randomized trials. Lancet2012, 37, 432–445.
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbing, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer. Nature2009, 462, 1005–1010.
- Russnes, H.; Navin, N.; Hieks, J.; Borresen-Dale, A.L. Insight into the heterogeneity of breast cancer through next-generation sequencing. Clin. Investig.2011, 121, 3810–3818.
- Miron, A.; Varadi, M.; Carrasco, D.; Li, H.; Luongo, L.; Kim, H.J.; Park, S.Y.; Cho, E.Y.; Lewis, G.; Kehoe, S.; et al. PI3KCA mutations in in situ and invasive breast carcinomas. Cancer Res.2010, 70, 5674–5678.
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Fredrick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and traslocations across breast cancer subtypes. Nature2012, 486, 405–409.
- Ellis, M.J.; Ding, L.I.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole genome analysis informs breast cancer response to aromatase inhibition. Nature2012, 486, 353–360.
- Curtis, C.; Shah, S.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature2012, 486, 346–352.
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, J.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature2012, 486, 400–405.
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell2012, 149, 979–993.
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshlal, J.; Ramakrishna, M.; et al. The life history of 21 breast cancers. Cell2012, 149, 994–1007.
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Cell2012, 46, 424–435.
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature2012, 490, 61–70.
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.; Tjan-Heijnen, V.C.; Labot, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Genet.2019, 51, 1450–1458.
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zon, X.; Matincorena, I.; Alexandrov, L.M.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature2016, 534, 47–53.
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.S.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Samaut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancer refines their genomic and transcriptomic landscapes. Commun.2016, 7, 11479.
- Griffith, O.L.; Spies, N.C.; Anurag, M.; Griffith, M.; Luo, J.; Tu, D.; Yeo, B.; Kunisaki, J.; Miller, C.A.; Krysiak, K.; et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Commun.2018, 9, 3476.
- Mosele, F.; Stefanovska, B.; Lusque, A.; Tran Dien, A.; Garberis, I.; Droin, N.; Le Tourneau, C.; Sablin, M.P.; Lacroix, L.; Enrico, D.; et al. Outcome and molecular landscape of patients with PI3CA-mutated metastatic breast cancer. Ann Oncol 2020, 31, 377-386.
- Zarvadas, D.; te Marveld, L.; Milne, R.L.; Fumagalli, D.; Fountzilas, G.; Kotoula, V.; Razis, E.; Papaxoinis, G.; Joensuu, H.; Moynahan, M.E.; et al. Tumor PIK3CA genotype and prognosis in early-stage breast cancer: a pooled analysis of individual patient data. Clin. Oncol. 2018, 38, 981-990.
- Luen, S.J.; Asher, R.; Lee, C.K.; Savas, P.; Kammler, R.; Dell’Orto, P.; Biasi, O.M.; Demanse, D.; LeBailey, L.; Dolan, S.; et al. Association of somatic driver alterations with prognosis in postmenopausal, hormone receptor-positive, HER2-negative early breast cancer: a secondary analysis of the BIG 1-98 randomized clinical trial. JAMA Oncol. 2018, 4, 1335-1343.
- Luen, S.J.; Asher, R.; Lee, C.K.; Savas, P.; Kammler, R.; Dell’Orto, P.; Biasi, O.M.; Demanse, D.; Hackl, W.; Theurlimann, B.; et al. Identifying drivers associated with increased risk of late distant recurrence in postmenopausal, estrogen receptor-positive, HER2-negative early breast cancer: results from the BIG 1-98 study. Oncol. 2020, 31, 1359-1368.
- Migliaccio, I.; Poali, M.; Risi, E.; Biagioni, C.; Biaganzoli, L.; Benelli, M.; Malorni, L. PIK3CA co-occurring mutations and copy-nimber gain in hormone receptor positive and HER2 negative breast cancer. NPJ Breast Cancer 2022, 8, 24.
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Genet.2013, 45, 1127–1133.
- Zack, D.I.; Schumacher, S.E.; Carter, S.L.; Chernick, A.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Genet.2013, 45, 1134–1140.
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, X.; Scheet, P.; Vattathils, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed single nucleus genome sequencing. Nature2014, 512, 155–160.
- Endesfelder, D.; Burrell, R.A.; Kanu, N.; McGranahan, N.; Howell, M.; Parker, P.J.; Downard, J.; Swanton, C.; Kschscho, M. Chromosomal instability selects gene copy-number variants encoding core regulators of proliferation in ER+breast cancer. Cancer Res. 2014, 74, 4853–4863.
- Zhang, L.; Feizi, N.; Chi, C.; Hu, P. Association analysis of somatic copy number alteration burden with breast cancer survival. Genet.2018, 9, 421.
- Hieronymous, H.; Musali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife2018, 7, e37294.
- Loibl, S.; Treue, D.; Budczies, J.; Weber, K.; Stenzinger, A.; Schmitt, W.D.; Weichert, W.; Jank, P.; Furlanetto, J.; Klauschen, F.; et al. Mutational diversity and therapy response in breast cancer: A sequencing analysis in the neoadjuvant GeparSepto trial. Cancer Res.2019, 25, 3986–3995.
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumors. Nature2000, 406, 747–752.
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Clin. Oncol.2009, 27, 1160–1167.
- Prat, A.; Cheang, M.C.; Galvan, P.; Nuciforo, P.; Paré, L.; Adamo, D.; Munoz, M.; Viladot, M.; Press, M.F.; Gagnon, R.; et al. Prognostic value of intrinsic subtypes in hormone receptor-positive metastatic breast cancer treated with letrazole with or without lapatinib. JAMA Oncol.2016, 2, 1287–1294.
- Cejalvo, J.M.; Martinez de Duenas, E.; Galvan, P.; Garcia-Recio, S.; Burgués Gasion, O.; Paré, L.; Antolin, S.; Martinello, R.; Blancas, I.; Adamo, B.; et al. Intrinsic subtypes and gene expression profiles in rpimary and metastatic breast cancer. Cancer Res.2017, 77, 2213–2221.
- Reis, P.P.; Waldron, L.; Goswami, R.S.; Xu, W.; Xuan, Y.; Perez-Ordonbez, B.; Gullane, P.; Irish, J.; Jurisica, I.; Karnal-Reid, S. mRNA transcript quantification in archival samples using multiplexed, color-coded probes. BMC Biotechnol.2011, 11, 46.
- Wallden, B.; Storhoff, J.; Nielsen, T.; Dowidar, N.; Schaper, C.; Ferre, S.; Liu, S.; Leung, S.; Geiss, G.; Snider, J.; et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med. Genom.2015, 8, 54.
- Picornell, A.C.; Echavarria, I.; Alvarez, E.; Lopez-Tarruella, S.; Jerez, Y.; Hoadley, K.; Parker, J.S.; Del Monte-Milian, M.; Ramos-Medina, R.; Gayarre, J.; et al. Breast cancer PAM50 signature: Correlation and concordance between RNA-Seq and digital multiplexed gene expression technologies in a triple negative breast cancer series. BMC Genom.2019, 20, 452.
- Kwa, M.; Makris, A.; Esteva, F.J. Clinical utility of gene-expression signatures in early stage breast cancer. Rev. Clin. Oncol.2017, 14, 595–610.
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Braehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. Engl. J. Med.2004, 35, 2187–2826.
- Dowsett, M.; Cuzick, J.; Wale, C.; Forbes, J.; Mallon, E.A.; Salter, J.; Quinn, E.; Dunbier, A.; Baum, M.; Buzdar, A.; et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrazole or tamoxifen: A TransATAC study. Clin. Oncol.2010, 28, 1829–1834.
- Buyse, M.; Loi, S.; van’t Veer, L.; Viale, G.; Delorenzi, M.; Glas, A.M.; Saghatchian d’Assignies, M.; Bergh, J.; Lidereau, R.; Ellis, P.; et al. Validation and clinical utility of a 70-gene prognostic signature in women with node-negative breast cancer. Natl. Cancer Inst.2006, 98, 1183–1192.
- Cardoso, F.; van’t Veer, J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.Y.; Brain, E.; Causeret, S.; DeLorenzi, M.; et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. Engl. J. Med.2016, 375, 717–729.
- Wuerstlein, R.; Kates, R.; Gluz, O.; Grischke, E.M.; Schem, C.; Thill, M.; Hasmueller, S.; Kohler, A.; Otremba, B.; Griesinger, F.; et al. Strong impact of MammaPrint and BluePrint on treatment decisions in luminal early breast cancer: Results of the WSG-PRIMe study. Breast Cancer Res. Treat.2019, 175, 389–399.
- Ali, H.R.; Rueda, O.M.; Chin, S.F.; Curtis, C.; Dunning, M.J.; Aparicio, S.; Caldas, C. Genome-driven integrated classification of breast cancer validated in over 7500 samples. Genome Biol.2014, 15, 431.
- Dawson, S.J.; Rueda, O.M.; Aparicio, S.; Caldas, C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J.2013, 32, 617–628.
- Mukherjee, A.; Russell, R.; Suet-Feung, C.; Liu, B.; Rueda, O.M.; Ali, H.R.; Turashvili, G.; Mahler-Araujo, B.; Ellis, I.O.; Aparicio, S.; et al. Associations between genomic stratification of breast cancer and centrally reviewed tumor pathology in the METABRIC cohort. NPJ Breast Cancer2018, 4, 5.
- Rueda, O.M.; Sammut, S.J.; Seaone, J.A.; Chin, S.F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of breast-cancer relapse reveal late-occurring ER-positive genomic subgroups. Nature2019, 567, 399–404.
- Liu, X.Y.; Ma, D.; Xu, X.E.; Jin, X.; Yu, K.D.; Jiang, Y.Z.; Shao, Z.M. Genomic landscape and endocrine resistant subgroup in estrogen receptor-positive, progesterone receptor-negative, and HER2-negative breast cancer. Theranotics2018, 8, 6386–6399.
- Ethier, J.L.; Ocana, A.; Rodriguez Lescure, A.; Ruiz, A.; Alba, E.; Calvo, L.; Ruiz-Borrego, M.; Santaballa, A.; Rodriguez, C.A.; Crespo, C.; et al. Outcomes of single versus double hormone receptor-positive breast cancer. A GEICAM/9906 sub-study. J. Cancer2018, 94, 199–205.
- Wu, N.; Fu, F.; Chen, L.; Lin, Y.; Yang, P.; Wang, C. Single hormone receptor-positive breast cancer patients experienced poor survival outcomes: A systematic review and meta-analysis. Transl. Oncol.2019, in press.
- Vallon-Christersson, J.; Hakkinen, J.; Hegardt, C.; Saal, L.H.; Larsson, C.; Ehinger, A.; Lindman, H.; Olofsson, H.; Sjoblom, T.; Warnberg, F.; et al. Cross comparison and prognostic assessment of breast cancer multigene signatures in a large population-based contemporary clinical series. Rep.2019, 9, 12184.
- Laibl, S.; Gianni, L. HER2-positive breast cancer. Lancet2017, 389, 2415–2429.
- Ross, J.S.; Gay, L.M.; Wang, K.; Ali, S.M.; Chumsri, S.; Elvin, J.A.; Bose, R.; Vergilio, J.A.; Suh, J.; Yelensky, R.; et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: an emerging opportunity for anti-HER2 targeted therapies. Cancer 2016, 122, 2654-2662.
- Kurozumi, S.; Alsaleem, M.; Monteiro, C.J.; Bharwaj, K.; Joosten, S.; Fujii, T.; Shirabe, K.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; et al Targetable ERR2 mutation status is an independent marker of adverse prognosis in estrogen receptor positive, ERBB2 non-amplified primary lobular breast carcinoma: a retrospective in silico analysis of public datasets. Breast Cancer Res 2020, 22, 85.
- Ross, J.S.; Wang, K.; Sheehan, C.E.; Boguniewicz, A.B.; Otto, G.; Dowing, S.R.; Sun, J.; He, J.; Curran, J.A.; Ali, S.; et al. Relapsed classic E-Cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res 2013, 19, 2668-2676.
- Ding, Q.; Chen, H.; Lim, B.; Damodaran, S.; Chen, W.; Tripathy, D.; Piha-Paul, S.; Luthra, R.; Meric-Bernstam, F.; Sahin, A.A. HER2 somatic mutation analysis in breast cancer: correlation with clinicopathological features. Human Pathol 2019, 92, 32-38.
- Yi, Z.; Rong, G.; Guan, Y.; Li, J.; Chang, L.; Li, H.; Liu, B.; Wang, W.; Guan, X.; Ouyang, Q.; Molecular landscape and efficacy of HER2-targeted therapy in patients with HER2-mutated metastatic breast cancer. NPJ Breast Cancer 2020, 6, 59.
- Zuo, W.J.; Yang, Y.Z.; Wang, Y.J.; Xu, X.E.; Hu, X.; Liu, G.Y.; Wu, J.; Di, G.H.; Yu, K.D.; Shao, Z.M. Dual characteristics of novel HAR2 kinase domain mutations in response to HER2-targeted therapies in human breast cancer. Clin Cancer Res 2016, 22, 4859-4869.
- Nayar, U.; Cohen, O.; Kapstad, C.; Cuoco, M.S.; Waks, A.G.; Wander, S.A.; Painter, C.; Freeman, S.; Persky, N.S.; Marini, L.; et al. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor-directed therapies. Nat Genet 2019, 51, 207-216.
- Medford, A.J.; Dubash, T.D.; Juric, D.; Spring, L.; Niemierko, A.; Vidula, N.; Peppercorn, J.; Isakoff, S.; Reeves, B.A.; LiCausi, J.A.; et al. Blood-based monitoring identifies acquired and targetable driver HER2 mutations in endocrine-resistant metastatic breast cancer. NPJ Precision Oncol 2019, 3,18.
- Ferrari, A.; Salomon, A.V.; Pivot, X.; Sertier, A.S.; Thomas, E.; Tenon, L.; Boyault, S.; Mulugeta, E.; Treilleux, I.; MacGrogan, G.; et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Commun.2016, 7, 12222.
- Daemen, A.; Manning, G. HER2 is not a cancer subtype but rather a pan-cancer event and is highly enriched in AR-driven breast tumors. Breast Cancer Res.2018, 20, 8.
- Zhao, S.; Liu, X.Y.; Jin, X.; Ma, D.; Xiao, Y.; Shao, Z.M.; Yiang, Y.Z. Molecular portraits and trastuzumab responsiveness of estrogen receptor-positive, progesterone receptor-positive, HER2-positive breast cancer. Theranotics2019, 9, 4935–4945.
- Chen, B.; Zhang, G.; Wei, G.; Wang, Y.; Guo, L.; Lin, J.; Li, K.; Mok, H.; Cao, L.; Ren, C.; et al. Heterogeneity of genomic profile in patients with HER2-posiitve breast cancer. Endocrine-Related Cancer 2020, 27, 153-162.
- Yap, Y.S.; Singh, A.P.; Lim, J.H.C.; Ahn, J.H.; Jung, K.H.; Kim, J.; Dent, R.A.; Ng, R.; Kim, S.B.; Chiang, D.Y. Elucidating therapeutic molecular targets in premenopausal Asian women with recurrent breast cancers. NPJ Breast Cancer2018, 4, 19.
- De Oliveira Taveira, M.; Nabavi, S.; Wang, Y.; Tonellato, P.; Esteva, F.J.; Cantley, L.C.; Wulf, G.M. Genomic characteristics of trastuzzumab-resistant Her2-positive metastatic breast cancer. J Cancer Res. Clin. Oncol.2017, 143, 1255–1262.
- Carey, L.A.; Berry, D.A.; Cirrincione, C.T.; Barry, W.T.; Pitcher, B.N.; Harris, L.N.; Ollila, D.W.; Krop, I.E.; Henry, N.L.; Weckstein, D.J.; et al. Molecular heterogeneity and response to neoadjuvant human epidermal growth factor receptor 2 targeting in CALGB 40601, a randomized phase III trial of paclitaxel plus trastuzumab with or without lapatinib. Clin. Oncol.2016, 34, 542–549.
- Liu, J.C.; Voisin, V.; Bader, G.D.; Deng, T.; Pusztai, L.; Symmans, W.F.; Esteva, F.J.; Egan, S.E.; Zacksenhaus, E. Seventeen-signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2+:ERα-breast cancer. Natl. Acad. Sci. USA 2012, 109, 5832–5837.
- Lesurf, R.; Griffith, O.L.; Griffith, M.; Hundal, J.; Trani, L.; Watson, M.A.; Aft, R.; Ellis, M.J.; Ota, D.; Suman, V.J.; et al. Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy results from the ACOSOG 21041 (Alliance) trial. Oncol.2017, 28, 1070–1077.
- Llombart-Cussac, A.; Cortés, J.; Paré, L.; Galván, P.; Bermejo, B.; Martínez, N.; Vidal, M.; Pernas, S.; López, R.; Muñoz, M.; et al. HER2-enriched subtype as a predictor of pathological complete response following trastuzumab and lapatinib without chemotherapy in early-stage HER2-positive breast cancer (PAMELA): An open-label, single-group, multicentre, phase 2 trial. Lancet Oncol.2017, 18, 545–554.
- Fumagalli, D.; Venet, D.; Ignatiadis, M.; Azim, H.A.; Maetens, M.; Rothé, F.; Salgado, R.; Bradbury, I.; Pusztai, L.; Harbeck, N.; et al. RNA sequencing to predict response to neoadjuvant anti-HER2 therapy: A secondary analysis of the NeoALTTO randomized clinical trial. JAMA Oncol.2017, 3, 227–234.
- Pernas, S.; Petit, A.; Climent, F.; Paré, L.; Perez-Martin, J.; Ventura, L.; Bergamino, M.; Galvan, P.; Falo, C.; Morilla, I.; et al. PAM50 subtypes in baseline and residual tumors following neoadjuvant trastuzumab-based chemotherapy in HER2-positive breast cancer: A consecutive-series from a single institution. Oncol.2019, 9, 707.
- Prat, A.; Pascual, T.; De Angelis, C.; Gutierrez, C.; Llombart-Cussac, A.; Wang, T.; Cortes, J.; Rexer, B.; Paré, L.; Forero, A.; et al. HER2-enriched subtype and ERBB2 expression in HER2-positive breast cancer treated with dual HER2 blockade. Natl. Cancer Inst.2020, 112, 46–54.
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldrisch, A.; de Azambuja, E.; Castro, G.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 years follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: Final analysis of the HERceptin adjuvant trial (HERA) trial. Lancet2017, 389, 1195–1205.
- Tolanev, S.M.; Guo, H.; Pernas, S.; Barry, W.T.; Dillon, D.A.; Ritterhouse, L.; Schneider, B.P.; Shen, F.; Fuhrman, K.; Baltay, M.; et al. Seven-year follow-up analysis of adjuvant paclitaxel and trastuzumab trial for node-negative, human epidermal growth factor receptor 2-positive breast cancer. Clin. Oncol.2019, 37, 1868–1875.
- He, X.; Ji, J.; Tian, M.; Esteva, F.J.; Hortobagyi, G.N.; Yesunbg, S.J. Long-term survival analysis of adjuvant chemotherapy with or without trastuzumab in patients with T1, node-negative HER2-positive breast cancer. Cancer Res.2019, in press.
- Von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Lintch, M.; Wodmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. Engl. J. Med.2019, 380, 817–828.
- Ma, F.; Ouyang, Q.; Li, W.; Jiang, Z.; Tong, Z.; Liu, Y.; Li, H.; Yu, S.; Feng, J.; Wang, S.; et al. Pyrotinib or lapatinib combined with capecitabine in HER2-positive metastatic breast cancer with prior taxanes, anthracyclines, and/or trastuzumab: A randomized, phase II study. Clin. Oncol.2019, in press.
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther.2019, 4, 34.
- Tanioka, M.; Fan, C.; Parker, J.S.; Hoadley, K.A.; Hu, Z.; Li, Y.; Hyslop, T.M.; Pitcher, B.N.; Soloway, M.G.; Spears, P.A.; et al. Integrated analysis of RNA and DNA from the phase III trial CALGB 40601 identifies predictors of response to trastuzumab -based neoadjuvant chemotherapy in HER2-positive breast cancer. Cancer Res.2018, 24, 5292–5304.
- Nuciforo, P.; Pascual, T.; Cortés, J.; Llombart-Cussac, A.; Fasani, R.; Paré, L.; Oliveira, M.; Galvan, P.; Martinez, N.; Bermejo, B.; et al. A predictive model of pathologic response based on tumor cellularity tumor-infiltrating lymphocytes (CelTIL) in HER2-positive breast cancer treated with chemo-free dual HER2 blockade. Oncol.2018, 29, 170–177.
- Di Cosimo, S.; Triulzi, T.; Pizzamiglio, S.; De Cecco, L.; De Azambuja, E.; Fumagalli, D.; Putzai, L.; Harbeck, N.; Izquierdo, M.; de la Pena, L.; et al. The 41-gene classifier TRAR predicts response of HER2 positive breast cancer patients in the NeoALTTO study. J. Cancer2019, 118, 1–9.
- Schettini, F.; Pascual, T.; Conte, B.; Chic, N.; Braso-Maristany, F.; Galvàn, P.; Martinez, O.; Adamo, B.; Vidal, M.; Munoz, M.; et al. HER2-enriched subtype and pathological complete response in HER2-positive breast cancer: A systematic review and meta-analysis. Cancer Treat. Rev.2020, 84, 101965.
- Gianni, L.; Bisagni, G.; Colleoni, M.; Del Mastro, L.; Zamagni, C.; Mausutti, M.; Zambetti, M.; Frassoldati, A.; De Fato, R.; Valagussa, P.; et al. Neoadjuvant treatment with trastuzumab and teruzumab plus palbociclib and fulvestrant in HER2-positive, ER-positive breast cancer (NA-PHER2): An exploratory, open-label, phase 2 study. Lancet2018, 19, 249–256.
- Ciruelas, E.; Villagrasa, P.; Paré, L.; Oliveira, M.; Pernas, S.; Cortés, J.; Soberino, J.; Adamo, B.; Vazquez, S.; Martinez, N.; et al. SOLTI-1303 PATRICIA phase II trial (STAGE1)- palbociclib and trastuzumab in postmenopausal patients with HER2-positive metastatic breast cancer. Cancer Res.2019, 79.
- Croessman, S.; Formisano, L.; Kinch, L.N.; Gonzalez-Ericsson, P.; Sudhan, D.R.; Nagy, R.J.; Matthew, A.; Bernicker, E.M.; Cristofanili, M.; He, J.; et al. Combined blockade of activating ERBB2 mutations and ER results in synthetic lethality of ER+/HER mutant breast cancer. Cancer Res.2019, 25, 278–289.
- Martin, M.; Holmes, F.A.; Elertsen, B.; Delaloge, S.; Moy, B.; Iwata, H.; von Minckiwitz, G.; Chia, G.; Mansi, T.; Barrios, C.H.; et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExtNET): 5-year analysis of a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol.2017, 18, 1688–1700.
- Unni, N.; Sudhan, D.R.; Artega, C.L. Neratinib: Inching up on the cure rate of HER2+breast cancer? Cancer Res. 2018, 24, 3483–3485.
- Ma, C.X.; Bose, R.; Gao, F.; Freedman, R.A.; Telli, M.L.; Kimmick, G.; Winer, E.; Naughton, M.; Goetz, M.P.; Russell, C.; et al. Neratinib efficacy and circulating tumor DNA detection of HER2 mutations in HER2 nonamplified metastatic breast cancer. Clin Cancer Res 2017, 23, 5687-5695.
- Smyth, L.M.; Piha-Paul, S.A.; Won, H.H.; Schram, A.M.; Saura, C.; Loi, S.; Lu, J.; Shapiro, G.I.; Juric, D.; Mayer, I.A.; et al. Efficacy and determinants of response to HER kinase inhibtion in HER2-mutatnt metastatic breast cancer. Cancer Discov 2020, 10, 198-213.
- Turner, N.C.; Kingston, B.; Kilburn L.S.; Kernaghan, S.; Wardley, A.M.; Macpherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct tehrapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol 2020, 21, 1296-1308.
- Untch, M.; von Minckiwitz, G.; Gerber, B.; Schem, C.; Rezai, M.; Fasching, P.A.; Tesch, H.; Eggermann, H.; Harmsch, C.; Huober, J.; et al. Trastuzumab or lapatinib in patients with human epidermal growth factor receptor 2-positive breast cancer in the GeparQuinto (G5) study (GBG 44). Clin. Oncol.2018, 36, 1308–1316.
- Sudhan, M.; Schwartz, L.J.; Guerrero-Zotano, A.; Formisano, L.; Nixon, M.J.; Croessmann, S.; Gonzalez-Ericsson, P.; Sauders, M.; Balko, J.M.; Avogadri-Connors, F.; et al. Extended adjuvant therapy with neratinib plus fulvestran block ER/HER2 crosstalk and maintains complete responses of ER+/HER2+breast cancers: Implications to the ExteNET trial. Cancer Res. 2019, 25, 771–783.
- Ma, C.X.; Luo, J.; Freedman, R.A.; Pluard, T.J.; Nangia, J.R.; Lu, J.; Valdez-Albini, F.; Cobleigh, M.; Jones, J.M.; Lin, N.U.; et al. The phase II MutHER study of neratinib alone and in combination with fulvestrant in HER2-mutated, non-apmplified metastatic breast cancer. Clin Cancer Res 2022, in press.
- Hanker, A.B.; Brewer, M.R.; Sheehan, J.H.; Koch, J.P.; Sliwoski, G.R.; Nagy, R.; Lanman, R.; Berger, M.F.; Hyman, D.M.; Solit, D.B.; et al. An acquired HER2T798I gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven breast cancer. Cancer Discovery 2017, 7, 575-585.
- Hanker, A.B.; Brown, B.P.; Meiler, J.; Marin, A.; Jayanthan, H.S.; Ye, D.; Lin, C.C.; Akamatsu, H.; Lee, K.M.; Chatterjee, S.; et al. Co-occurring gain-of-function mutations in HER2 and HER3 modulate HER2/HER3 activation, oncogenesis, and HER2 inhibitor sensitivity. Cancer Cell 2021, 39, 1099-1114.
- Smith, A.E.; Ferraro, E.; Safonov, A.; Morales, C.B.; Arenas Lahuerta, E.J.; Li, Q.; Kulick, A.; Ross, D.; Solit, D.B.; de Stanchina, E.; et al. HER2+ breast cancers evade anti-HER2 therapy via a switch in driver pathway. Nat Commun 2021, 12, 6667.
- Chiu, C.G.; Masoudi, H.; Leung, S.; Voduk, D.K.; Gilks, B.; Huntsman, D.G.; Wiseman, S.M. HER-3 overexpression is prognostic of reduced breast cancer survival: a study of 4046 patients. Ann Surg 2010, 251, 1107-1116.
- Diwanji, D.; Trenker, R.; Thaker, T.M.; Wang, F.; Agard, D.A.; Verba, K.A.; Jura, N. Structures of the HER2-HER3-NRG1β complex reveal a dynamic interface. Nature 2021, 600, 339-343.
- Campbell, M.R.; Ruiz-Saenz, A.; Zhang, Y.; Peterson, E.; Steri, V.; Oeffinger, J.; Sampang, M.; Jura, N.; Moasser, M.M. Extensive conformational and physical plasticity protects HER2-HER3 tumorigenic signaling. Cell Reports 2022, 38, 110285.
- Campbell, M.R.; Ruiz-Saenz, A.; Peterson, E.; Agnew, C.; Ayaz, P.; Garfinle, S.; Littlefield, P.; Steri, V.; Oeffinger, J.; Sampang, M.; et al. Targetable HER3 functions driving tumorigenic signaling in HER2-amoplified cancers. Cell Reports 2022, 38, 110291.
- Chang, C.A.; Jen, J.; Jiang, S.; Sayad, A.; Mer, A.S.; Brown, K.R.; Nixon, A.; Dhabaria, A.; Tang, K.H.; Venet, D.; et al. Ontogeny and vulnerabilities of drug-tolerant persisters in HER2+ breast cancer. Cancer Discov 2022, in press.
- Gandullo-Sanchez, L.; Capone, E.; Ocana, A.; Iacobelli, S.; Sala, G.; Pandiella, A. HER3 targeting with an antibody-drug conjugate bypasses resistance to anti-HER2 therapies. EMBO Mol Med 2020, 12, e11498.
- Odintsov, I.; Lui, A.; Sisso, W.; Gladstone, E.; Liu, Z.; Delasos, L.; Kurth, R.; Sisso, E.M.; Vojnic, M.; Khodos, I.; et al. The anti-HER3 mAb serimantumab effectively inhibits growth of patient-derived and isogenic cell line and xenograft models with oncogenic NRG1 fusions. Clin Cancer Res 2021, 27, 3154-3166.
- Seung, E.; Xing, Z.; Wu, L.; Rao, E.; Cortez-Ratamozo, V.; Ospina, B.; Chen, L.; Beil, C.; Song, Z.; Zhang, B.; et al. A trispecific antibody targeting HER2 and T cells inhibits breast cancer growth via CD4 cells. Nature 2022, in press.
- Early Breast Cancer Trialists’ Collaborative group. Trastuzumab for early-stage, HER2-positive breast cancer: a meta-analysis of 13864 women in seven randomized trials. Lancet Oncol 2021, 22, 1139-1150.
- Rakha, E.A.; Elsheikh, S.E.; Aleskandarany, M.A.; Habashi, H.O.; Green, A.R.; Powe, D.G.; El-Sayed, M.E.; Benhasouna, A.; Brunet, J.S.; Akslen, L.A.; et al. Triple-negative breast cancer: Distinguishing between basal and nonbasal subtypes. Cancer Res.2009, 15, 2302–2310.
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123-133.
- Rakha, E.A.; Allison, K.H.; Ellis, I.O.; Horri, D.; Masuda, S.; Penault-Llorca, F.; Tsuda, H.; Vincent-Salomon, A. Invasive breast carcinoma: general overview. In: WHO Classification of Tumors Editorial Board, eds. WHO Classification of Tumours, 5th, Breast Tumors. IARC Press, Lyon, 2019, pp. 82-101.
- Rakha, E.A.; Allison, K.H.; Bu, H.; Ellis, I.O.; Foschini, M.P.; Horii, R.; Masuda, S.; Penault-Llorca, F.; Schnitt, S.J.; Tsuda, H.; et al. Invasive breast carcinoma of no special type. WHO Classification of Tumors Editorial Board, eds. WHO Classification of Tumours, 5th, Breast Tumors. IARC Press, Lyon, 2019, pp. 102-109.
- Garrido-Castro, A.; Lin, N.U.; Polyak, K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection of treatment. Cancer Discovery 2019, 9, 176-198.
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Glicrease, M.Z.; Krishanmurthy, S.; Lee, J.S.; Fridyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res.2009, 69, 4116–4124.
- Taube, J.H.; Herschckowitz, J.L.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Ondet, T.; Gupta, P.B.; Evans, K.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Natl. Acad. Sci. USA2010, 107, 15449–15454.
- Prat, A.; Parker, J.; Karginova, O.; Fan, C.; Livasy, C.; I Herschkowitz, J.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res.2010, 12, R68.
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sorlie, T. Re-definition of claudin-low as a breast cancer phenotype. Nat Commun 2020; 11: 1787.
- Pommier, R.M.; Sanlaville, A.; Tonon, L.; Kielbassa, J.; Thomas, E.; Ferrari, A.; Sertier, A.S.; Hollande, F.; Martinez, P.; Tissier, A.; et al. Comprehensive characterization of claudin-low breast tumors reflects the impact of the cell-of-origin on cancer evolution. Nat Commun 2020, 11, 3431.
- Radler, P.D.; Wehde, B.L.; Triplett, A.A.; Shrestha, H.; Shephjerd, J.H.; Pfefferle A.D.; Rui, H.; Cardiff, R.D.; Perou, C.M.; Wagner, K.U. Highly metastatic claudin-low mammary cancers can originate from luminal epithelial cells. Nat Commun 2021, 12, 3742.
- Tang, Y.H.; Rockstroh, A.; Sokolowski, K.A.; Lynam, L.R.; Lehman, M.; Thompson, E.W.; Gregory, P.A.; Nelson, C.C.; Volpert, M.; Hollier, B.G. Neuropilin-1 is over-expressed in claudin-low breast cancer and promotes tumor progression through acquisition of stem cell characteristics and RAS/MAPK pathway activation. Breast Cancer Res 2022, 24,8.
- Balkenhol, M.; Vreuls, W.; Wauters, C.; Mol, S.; van der Laak, J.; Bult, P. Histological subtypes in triple negative breast cancer are associated with specific information on survival. Ann Diagn Pathol 2020, 46, 151490.
- Bando, Y.; Kobayashi, T.; Miyakami, Y.; Sumida, S.; Kakimoto, T.; Saijo, Y.; Uehara, H. Triple-negative breast cancer and basal-like subtype: pathology and targeted therapy. J Med Investig 2021, 68, 213-219.
- Pareja, F.; Geyer, F.C.; Marchiò, C.; Burke, K.A.; Weigedt, B.; Reis-Filho, J.S. Triple-negative breast cancer: the importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer 2016, 2, 16036.
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature2012, 486, 395–399.
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. Clin. Investig.2011, 121, 2750–2767.
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, R.D.; Pieteropal, A.J.; Hetobagyi, G.V.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Cancer Res.2013, 19, 5533–5540.
- Kriegsmann, M.; Endris, V.; Wolf, T.; Pfarr, N.; Stenzinger, A.; Loibl, S.; Denkert, C.; Schneeweiss, A.; Budczies, J.; Sinn, H.-P.; et al. Mutational profiles in triple-negative breast cancer defined by ultradeep multigene sequencing show high rates of PI3K pathway alterations and clinically relevant entity subgroup specific differences. Oncotarget2014, 5, 9952–9965.
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.; Contreras, A.; Fuqua, S.; Savage, M.; Osborne, K.; Hilsenbeck, S.; Chang, J.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Cancer Res.2014, 21, 1688–1698.
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.L.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE2016, 11, e0157368.
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; YU, Y.; et al. Genomic and transcriptomic landscape of triple-negative breast cancers: Subtypes and treatment strategies. Cancer Cell2019, 35, 428–440.
- Echavarria, I.; Lopez-Tarruela, S.; Picornell, A.; Garcia-Saenz, J.A.; Jerez, J.; Hoadley, K.; Gomez, H.L.; Moreno, F.; Monte-Millan, M.D.; Marquez-Rodas, I.; et al. Pathological response in triple-negative breast cancer cohort treated with neoadjuvant carboplatin and docetaxel according to Lehmann’s refined classification. Cancer Res.2018, 24, 1845–1852.
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Oncol.2018, 29, 895–902.
- Quist, J.; Mirza, H.; Cheang, M.; Telli, M.; O’Shaughnnessy, J.; Lord, C.; Tutt, A.; Grigoriadis, A. A four-gene decision tree signature classification of triple-negative breast cancer: Implications for targeted therapeutics. Cancer Ther.2019, 18, 204–212.
- Hsu, H.M.; Chu, C.M.; Chang, Y.J.; Yu, J.C.; Chen, C.T.; Jian, C.E.; Lee, C.Y.; Chiang, C.W.; Chang, Y.T. Six novel immunoglobulin genes as biomarkers for better prognosis in triple-negative breast cancer by gene co-expression network analysis. Rep.2019, 9, 4484.
- Jiang, Y.Z.; Liu, Y.; Xiao, Y.; Hu, X.; Jiang, L.; Zuo, W.J.; Ma, D.; Ding, J.; Zhu, X.; Zou, J.; et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic rtiple-negative breast cancer: the FUTURE trial. Cell Research 2021, 31, 178-186.
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanism of triple-negative breast cancer. Cancer Res.2019, 25, 5002–5014.
- Prado-Vazquez, G.; Gamez-Pozo, A.; Trilla-Fuertes, L.; Arevaliullo, J.M.; Zapater-Moros, A.; Ferrer-Gomez, M.; Diaz-Almiron, M.; Lopez-Vacas, R.; Navarro, H.; Main, P.; et al. A novel approach to triple-negative breast cancer molecular classification reveals a luminal immune-positive subgroup with good prognoses. Rep.2019, 9, 1538.
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwartz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sanchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov.2014, 4, 232–245.
- Jiang, T.; Shi, W.; Wali, V.B.; Pongor, L.S.; Li, C.; Lau, R.; Gyorfly, B.; Lifton, R.P.; Symmans, W.F.; Punztai, L.; et al. Predictors of chemosensitivity in triple negative breast cancer: An integrated genomic analysis. PLOS Med.2016, 13, e1002193.
- Lehmann, B.D.; Colaprico, A.; Silva, T.C.; Chen, J.; An, H.; Ban, Y.; Huang, H.; Wang, L.; James, J.L.; Balko, J.M.; et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat Commun 2021, 12, 6276.
- Santonja, A.; Sanchez-Munoz, A.; Liuch, A.; Chica-Parrado, M.R.; Albanell, J.; Chacon, J.I.; Antolin, S.; Jerez, J.M.; de la Haba, J.; de Luque, V.; et al. Triple negative breast cancer subtypes and pathologic complete response rate to neoadjuvant chemotherapy. Oncotarget 2018, 9, 26406-26416.
- Lehmann, B.D.; Abramson, V.G.; Sanders, M,E.; Mayer, E.L.; Mayer, E.L.; Haddad, T.C.; Nanda, R.; van Poznak, C.; Storniolo, A.M.; Nangia, J.R.; et al. TBCRC 032 IB/II multicenter study: molecular insights to AR antagonist and PI3K inhibitor efficacy in patients with AR+ metastatic triple-negative breast cancer. Clin Cancer Res 2020, 26, 2111-2123.
- Wang, D.Y.; Gendoo, D.; Ben-David, Y.; Woodgett, J.R.; Zackenhaus, E. A subgroup of microRNAs defines PTEN-deficient, triple-negative breast cancer patients with poorest prognosis and alterations in RB1, MYC, and Wnt signaling. Breast Cancer Res 2019, 21, 18.
- Wang, D.Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zackenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Scient Rep 2019, 9, 19107.
- Asleh, K.; Negri, G.L.; Spencer Miko, S.E.; Colborne, SV.; Hughes, C.S.; Wang, X.Q.; Gao, D.; Gilks, C.B.; Chia, S.; Nielsen, T.O.; et al. Proteomic analysis of archival breast cancer clinical specimens identifies biological subtypes with distinct survival outcomes. Nat Commun 2022, 13, 896.
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-negative breast cancer risk identified by multigene hereditary cancer panel testing. Natl. Cancer Inst.2018, 110, 855–862.
- Hu, C.; Polley, E.C.; Yadav, S.; Lilyquist, J.; Shimelis, H.; Na, J.; Hart, S.N.; Goldgar, D.E.; Shah, S.; Pesaran, T.; et al. The contribution of germline predisposition gene mutations to clinical subtypes of invasive breast cancer from a clinical genetic testing. J Natl Cancer Inst 2020, 112, djaa023.
- Zhang, H.; Ahern, T.U.; Lecarpentier, J.; Barnes, D.; Beesley, J.; Qi, G.; Jiang, X.; O’Mara, T.; Zhao, N.; Bolla, M.K.; et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat Genet 2020, 52, 572-581.
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A population-based study of genes previously implicated in breast cancer. N Engl J Med 2021, 384, 440-451.
- Dorling, L.; Carvalho, S.; Allen, J.; Gonzalez-Neira, A.; Luccarini, C.; Wahlstrom, C.; Pooley, K.A.; Parson, M.T.; Fortuno, C.; Wang, Q.; et al. Breast cancer risk genes – association analysis in more than 113,000 women. N, Engl J Med 2021, 384, 428-439.
- Mavaddat ; Dorling, L.; Cravalho, S.; Allen, J.; Gonzalez-Neira, A.; Keeman, R.; Bolla, M.K.; Dennis, J.; Wang, Q.; Ahearn, T.; et al. Pathology of tumors associated with pathogenic germline variants in 9 breast cancer susceptibility genes. JAMA Oncol. 2022, in press.
- Engel, C.; Rhiem, K.; Hahnen, E.; Loibl, S.; Weber, K.E.; Seller, S.; Zachariae, S.; Hauke, J.; Wappenschmidt, B.; Waha, A.; et al. Prevalence of pathogenic BRCA 1-2 germline mutations among 802 women with unilateral triple-negative breast cancer without family cancer history. BMC Cancer2018, 18, 265.
- Chen, H.; Wu, J.; Zhang, Z.; Tang, Y.; Li, X.; Liu, S.; Cao, S.; Li, X. Association between BRCA status and triple-negative breast cancer: A meta-analysis. Pharmacol.2018, 9, 909.
- Ellsworth, D.L.; Turner, C.E.; Ellsworth, R.E. A review of the hereditary component of triple negative breast cancer: High- and moderate-penetrance breast cancer genes, low-penetrance loci, and the role of nontraditional genetic elements. Oncol.2019, 4382606.
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresent-Dale, A.L.; Boyault, S.; et al. Signatures of mutational processes in human cancer. Nature2013, 500, 415–421.
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kubler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Genet.2017, 49, 1476–14876.
- Daviues, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Med.2017, 23, 517–525.
- Nones, K.; Johnson, J.; Newell, F.; Patch, A.M.; Thorne, H.; Kazakoff, S.H.; de Luca, X.M.; Parsons, M.T.; Ferguson, K.; Reid, L.E.; et al. Whole-genome sequencing reveals clinically relevant insights into the etiology of familial breast cancers. Oncol.2019, 30, 1071–1079.
- Staaf, J.; Glodzik, D.; Boch, A.; Vallon-Christersson, J.; Reutersward, C.; Hakkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-genome sequencing of triple-nagative breast cancers in a population-based clinical study. Med.2019, 25, 1526–1533.
- Sun, J.; Meng, H.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; et al. Germline mutations in cancer susceptibility genes in a large series of unselected breast cancer patients. Clin Cancer Res 2017, 23, 6113-6119.
- Annunziato, S.; de Ruiter, J.; Henneman, L.; Brabrillasca, C.S.; Lutz, C.; Vaillant, F.; Ferrante, F.; Drenth, A.P.; van der Burg, E.; Siteur, B.; et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat Commun 2019, 10, 397.
- Sun, S.; Brazhnil, K.; Lee, M.; Maslov, A.Y.; Zhang, Y.; Huang, Z.; Klugman, S.; Park, B.H.; Vijg, J.; Montagna, C. Single-cell analysis of somatic mutation burden in mammary epithelial cells of pathogenic BRCA1/2 mutation carriers. J Clin Invest 2022, 132, e148113.
- Yang, X.; Leslie, G.; Doroszuk, A., Schneider S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J Clin Oncol 2019, 38, 674-685.
- Li, A.; Geyer, F.C.; Blecua, P.; Lee, J.Y.; Selenica, P.; Brown, D.N.; Pareja, F.; Lee, S.; Kumar, R.; Rivera, B.; et al. Homologous recombination DNA repair defects in PALB-associated breast cancers. NPJ Breast Cancer 2019, 5, 23.
- Ng, P.S.; Pan, J.W., Zabidi, M.; Rajadurai, P.; Yip, C.H.; Reuda, O.M.; Dunning, A.M.; Antoniou, A.C.; Esaton, D.F.; Caldas, C.; et al. Characterisation of PALB2 tumours through whole-exome and whole-transcriptome analyses. NPJ Braest Cancer 2021, 17,46.
- Lee, J.; Li, N.; Rowley, S.M.; Cheasley, D.; Zethoven, M.; McInerny, S.; Gorringe, K.L.; James, P.A.; Campbell, I.G. Molecular analysis of PALB2-assocaited breast cancers. J Pathol 2018, 245, 53-60.
- Hu, Z.Y.; Liu, L.; Xie, N.; Lu, J.; Liu, Z.; Tang, Y.; Wang, Y.; Yang, J.; Ouyang, Q. Germline PALB2 mutations in cancers and its distinction from somatic PALB2 mutations in breast cancers. Front Genet 2020, 11, 829.
- Mandelker, D.; Kumar, R.; Pei, X.; Selenica, P.; Setton, J.; Arunalachalam, S.; Ceyhan-Birsoy, O.; Brown, D.N.; Norton, L.; Robson, M.E.; et al. The landscape of somatic genetic alterations in breast cancers from CHEK2 germline mutation carriers. J NCI Cancer Spectrum 2019, 3, pkz027.
- Wei, G.; Teng, M.; Rosa, M.; Wang, X. Unique ER PR expression pattern in breast cancers with CHEK2 mutation: a hormone receptor and HER2 analysis based on germline cancer predisposition genes. Breast Cancer Res 2022, 24, 11.
- Boonen, R.; Wiegant, W.W.; Celosse, N.; Vroling, B.; Heijl, S.; Kote-Jarai, Z.; Mijuskovic, M.; Cristea, S.; Solleveld-Westerink, N.; van Wezel, T.; et al. Functional analysis identifies damaging CHEK2 missense variants associated with increased cancer risk. Cancer Res 2022, 82, 615-631.
- Iyevleva, A.G.; Aleskakhina, S.N.; Sokolenko, A.P.; Baskina, S.V.; Venina, A.R.; Anisimova, E.I.; Bizin, I.V.; Ivantsov, A.O.; Belysheva, Y.V.; Chernyakova, A.P.; et al. Somatic loss of the remaining allele occurs approximately in half of CHEK2-driven breast cancers and is accompanied by a border-line increase of chromosomal instability. Breast cancer Res Treat 2022, 192, 283-291.
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline pathogenic variants in the ataxia teleangectasia mutated (ATM) gene are associated with high and moderate risks for multiple cancers. Cancer Prev Res 2021, 14, 433-440.
- Moslemi, M.; Moradi, Y.; Degghanbanadaki, H.; Afkhami, H.; Khaledi, M.; Sedighimehr, N.; Fathi, J.; Sohrabi, E. The association between ATM variants and risk of breast cancer: a systematic review and meta-analysis. BMC Cancer 2021, 21, 27.
- Toss, A.; Tenedini, E.; Piombino, C.; Venturelli, M.; Marchi, I.; Gasparini, E.; Brabieri, E.; Razzaboni, E.; Domati, F.; Caggia, F.; et al. Clinicopathologic profile of breast cancer in germline ATM and CHEK2 mutation carriers. Genes 2021, 12, 616.
- Renault, A.L.; Mebirouk, N.; Fuhrmann, L.; Bataillon, G.; Cavaciuti, E.; Le Gal, D.; Girard, E.; Popova, T.; La Rosa, P.; Beauvallet, J.; et al. Morphology and genomic hallmarks of breast tumours developed by ATM deleterious variant carriers. Breast Cancer Res 2018, 20, 28.
- Weigelt, B.; Bi, R.; Kumar, R.; Blecua, P.; Mendelker, D.L.; Geyer, F.C.; Pareja, F.; James, P.A.; ConFab Investigators, Couch, F.J.; Eccles, D.M.; et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. JNCI Journal 2018, 110, djy028.
- Waks, A.G.; Kim, D.; Jain, E.; Snow, C.; Kirkner, G.I.; Rosenberg, S.N.; Oh, C.; Poorvu, P.D.; Ruddy, K.I.; Tamimi, R.M.; et al. Somatic and germline genomic alterations in very young women with breast cancer. Clin Cancer Res 2022, in press.
- Andrikoupoulou, A.; Chatzinikolaou, S.; Kyriopoulou, I.; Bletsa, G.; Kaparelou, M.;Liontos, M.; Dimopoulos, M.A.; Zagouri, F. The mutational landscape of early-onset breast cancer: a next-generation sequencing analysis. Front Oncol 2022, 11, 797505.
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallassi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Cancer Res.2016, 22, 3764–3773.
- Robson, M.; Im, S.A.; Senkus, E. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. Engl. J. Med.2017, 377, 523–533.
- Senkus-Konefka, E.; Domchek, S.M.; Im, S.A.; Xu, B.; Armstrong, A.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Conte, P.; et al. 101 (PB-002): Subgroups analysis of olaparib monotherapy versus chemotherapy by hormone receptor and BRCA mutation status in patients with HER2-negative metastatic breast cancer and a germline BRCA mutation: OlympiAD. J. Cancer2018, 92, S19–S20.
- Eiermann, W.; Rugo, H.S.; Diab, S. Analysis of germline BRCA 1-2 mutated (gBRC mut) hormone receptor-positive (HR+) and triple negative breast cancer (TNBC) treated with talazoparib (TALA). Clin. Oncol.2018, 36, 1070.
- Litton, J.K.; Rugo, H.S.; Ettl, J. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. Engl. J. Med.2018, 379, 753–763.
- Blum, J.L.; Laird, A.D.; Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Martin, M.; Roche, H.H.; Lee, K.H.; Goodwin, A.; et al. Determinants of response to talazoparib in patients with HER2-negative, germline BRCA1/2-mutated breast cancer. Clin Cancer Res 2022, 28, 1-8.
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol.2019, 5, 1132.
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Domodaran, S.; De Snyder, S.M.; Brewster, A.M.; Barcenors, C.H.; Whitman, G.J.; et al. Neoadjuvant talazoparib for patients with operable breast cancer with a germline BRCA pathogenic variant. Clin. Oncol.2020, 38, 388–394.
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Grazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Berrett-Lee, P.; et al. Carboplatin in BRCA 1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT trial. Med.2018, 24, 628–637.
- De Talhouet, S.; Peron, J.; Vuilleumier, A.; Friedlaender, A.; Viassolo, V.; Ayme, A.; Bodmer, A.; Treilleux, I.; Lang, N.; Tille, J.C.; et al. Clinical outcome of breast cancer in carriers of BRCA1 and BRCA2 mutations according to molecular subtypes. Scient Rep 2020, 10, 7073.
- Loibl, S.; O’Shaughnessy, J.; Unch, M.; Sikoiv, V.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomized, phase 3 trial. Lancet Oncol.2018, 19, 497–507.
- Metzger-Filho, O.; Collier, K.; Asad, S.; Ansell, P.J.; Watson, M.; Bae, J.; Cherian, M., O’Shaughnessy, J.; Untch, M.; Rugo, H.S.; et al. Matched cohort study of germline BRCA mutation carriers with triple negative breast cancer in brightness. NPJ Breast Cancer 2021, 7, 142.
- Geyer, C.E.; Sikov, W.M.; Huober, J.; Rugo, H.S.; Wolmark, N.; O’Shaughnessy, J.; Maag, D.; Untch, M.; Golshan, M.; Lorenzo, J.P.; et al. Long-term efficacy and safety of addition of carboplatin with or without veliparib to standard neodjuvant chemotherapy in triple-negative breast cancer: 4-year follow-up data from BrighTNess, a randomized phase III trial. Ann Oncol 2022, in press.
- Shepherd, J.H.; Ballman, K.; Polley, M.Y.; Campbell, J.D.; Fan, C.; Selitsky, S.; Fernandez-Martinez, A.; Parker, J.S.; Hoadley, K.A.; Hu, Z.; et al. CALBG 400603 (Alliance): long-term outcomes and genomic correlates outcomes and genomic correlates of response and survival fater neoadjuvant chemotherapy with or without carboplatin and bavicizumab in triple-negative breast cancer. J Clin Oncol 2022, in press.
- Hancock, B.; Chen, Y.S.; Solzak, J.P.; Ahmad, M.N.; Wedge, D.C.; Brinza, D.; Scafe, C.; Veitch, J.; Gottimukkala, R.; Short, W.; et al. Profiling molecular regulators of recurrence in chemorefractory triple-negative breast cancers. Breast Cancer Res.2019, 21, 87.
- Gao, R.; Davis, A.; MacDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Genet.2016, 48, 1119–1130.
- Kim, C.; Gao, R.; Sei, E.; Brand, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell2018, 173, 879–893.
- Dalenc, F.; Lusque, A.; De La Motte Rouge, T.; Pistilli, B.; Brain, E.; Pasquier, D.; Debled, M.; Thery, J.C.; Goncalves, A.; Desmoulins, I.; et al. Impact of lobular versus ductal histology on overall survival in metastatic breast cancer: a French retrospective multicentre cohort study. Eur J Cancer 2022, 164, 70-79.
- Gamallo, C.; Palacios, J.; Suarez, A.; Pizarro, A.; Navarro, P.; Quintanilla, M.; Cano, A. Correlation of E-cadherin expression with differentiation grade and histological type in breast carcinoma. J. Pathol. 1993, 142, 987–993. Med. Sci.
- Dabbs, D.J.; Schnitt, S.J.; Geyer, F.C.; Weigelt, B.; Cachner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Lakhani, S.R.; et al. Lobular neoplasia of the breast revisited with emphasis on the role of E-cadherin immunohistochemistry. J. Surg. Pathol. 2013, 37, e1–e11.
- Sarrio, D.; Moreno-Bueno, C.; Hardisson, D.; Sanchez-Estevez, C.; Guo, M.; Herman, G.J.; Gamallo, G.; Esteller, M.; Palacios, J. Epigenetic and genetic alterations of APC and CDH1 genes in lobular breast cancer: Relationships with abnormal E-cadherin and catenin expression and microsatellite instability. J. Cancer 2003, 106, 208–215.
- Grabenstetter, A.; Mohanty, A.S.; DeLair, D.F.; Tan, L.K.; Ross, D.S. Correlation of CDH1 alterations and aberrant E-cadherin expression in lobular carcinomas. Cancer Res. 2019.
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519.
- Desmedt, C.; Zoppoli, G.; Gundem, G.; Pruneri, G.; Larsimont, D.; Fornili, M.; Fumagalli, D.; Brown, D.; Rothe, F.; Vincent, D.; et al. Genomic characterization of primary invasive lobular breast cancer. Clin. Oncol. 2016, 34, 1872–1881.
- Pareja, F.; Ferrando, L.; Lee, S.; Beca, F.; Selenica, P.; Brown, D.; Farmanbar, A.; Da Cruz Paula, A.; Vahdatinia, M.; Zhang, H.; et al. The genomix landscape of metastatic histologic special types of invasive breast cancer, NPJ Breast Cancer 2020, 6, 53.
- Sokol, E.S.; Feng, Y.X.; Jin, D.X.; Basudan, A.; Lee, A.V.; Atkinson, J.M.; Chen, J.; Stephens, P.J.; Frampton, G.M.; Gupta, P.B.; et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann Oncol 2019, 30, 115-123.
- Bergeron, A.; MacGrogan, G.; Bertaut, A.; Ladoire, S.; Arveux, P.; Desmoulins, I.; Bonnefoi, H.; Loustalot, C.; Auriol, S.; Beltjens, F.; et al. Triple-negative breast lobular carcinoma: a luminal androgen receptor carcinoma with specific ESRRA mutations. Modern Pathol 2021, 34, 1282-1296.
- Riedlinger, G.M.; Joshi, S.; Hinshfield, K.M.; Barnard, N.; Ganesan, S. Targetable alterations in invasive pleomorphic lobular carcinoma of the breast. Breast Cancer Res 2021, 23, 7.
- Yadav, S.; Hu, C.; Natahnson, K.L.; Weitzel, J.N.; Goldgar, D.E.; Kraft, P.; Gnanaolivu, R.D.; Na, J.; Huang, H., Boddicker, N.J.; et al. Germline pathogenic variants in cancer predisposition genes among women with invasive lobular carcinoma of the breast. J Clin Oncol 2021, 39, 3918-3926.
- Ng, C.K.Y.; Piscuoglio, S.; Geyer, F.C.; Burke, K.A.; Pareja, F.; Eberle, C.A.; Lim, R.S.; Natrajan, R.; Riaz, N.; Mariani, O.; et al. The landscape of somatic genetic alterations in metaplastic breast carcinomas. Cancer Res.2017, 23, 3859–3870.
- Ross, J.S.; Badve, S.; Wang, K.; Sheehan, C.E.; Boguniewicz, A.B.; Otto, G.A.; Yelensky, R.; Lipson, D.; Ali, S.; Morosini, D.; et al. Genomic profiling of advanced-stage, metaplastic breast carcinoma by next-generation sequencing reveals frequent, targetable abnormalities and potential new treatment options. Pathol. Lab. Med.2015, 139, 642–649.
- Joneja, U.; Vranic, S.; Swensen, J.; Feldman, R.; Chen, W.; Kimbrough, J.; Xiao, N.; Xiao, N.; Reddy, S.; Palazzo, J.; et al. Comprehensive profiling of metaplastic breast carcinomas reveals frequent overexpression of programmed death-ligand 1. Clin. Pathol.2017, 70, 255–259.
- Eldenfield, J.; Schammel, C.; Collins, J.; Schammel, D.; Edenfield, W.J. Metaplastic breast cancer: Molecular typing and identification of potential targeted therapies at a single institution. Breast Cancer2017, 17, e1–e10.
- Zhai, J.; Giannini, G.; Ewalt, M.D.; Zhang, E.Y.; Invernizzi, M.; Niland, J.; Lai, L.L. Molecular characterization of metaplastic breast carcinoma via next-generation sequencing. Pathol.2019, 86, 85–92.
- Gonzalez-Martinez, S.; Perez-Miles, B.; Carretero-Barrio, I.; Palacios-Barrequero, M.L.; Perez-Garcia, J.; Cortés, J.; Palacios, J. Molecular features of metaplastic breast carcinoma: an infrequent subtype of triple negative breast carcinoma. Cancers 2020, 12, 1832.
- Da Silva, E.M.; Selenica, P.; Vahdatinia, M.; Pareja, F.; Da Cruz Paula, A.; Ferrando, L.; Gazzo, A.M.; Dopeso, H.; Ross, D.S.; Bakhteri, A.; et al. TERT promoter hotspot mutations and gene amplification in metaplastic breast cancer. NPJ Breast Cancer 2021, 7, 43.
- Krings, G.; Chen, Y.Y. Genomic profiling of metaplastic breast carcinomas reveals genetic heterogeneity and relationship to ductal carcinoma. Pathol.2018, 31, 1661–1674.
- Dave, B.; Gonzalez, D.D.; Liu, Z.B.; Li, X.; Wong, H.; Granados, S.; Ezzedine, N.E.; Sieglaff, D.H.; Ensor, J.E.; Miller, K.D.; et al. Role of RPL39 in metaplastic breast cancer. Natl. Cancer Inst.2017, 109, 292.
- Afkhami, M.; Schmolze, D.; Yost, S.E.; Frankel, P.K.; Dagis, A.; Amanam, I.U.; Telatar, M.; Nguyen, K.; Yu, K.W.; Luu, T.; et al. Mutation and immune profiling of metaplastic breast cancer: Correlation with survival. PLoS ONE2019, 14, e0224726.
- McCart Reed, A.E.; Kalaw, E.; Nones, K.; Bettington, M.; Lim, M.; Bennett, J.; Johnstone, K.; Kutasovic, J.R.; Saunus, J.M.; Kazakoff, S.; et al. Phenotypic and molecular dissection of metaplastic breast cancer and the prognostic implications. Pathol.2019, 247, 214–227.
- Bartels, S.; van Luttikuizen, J.L.; Christgen, M.; Magel, L.; Luft, A.; Hanzelmann, S.; Lehmann, U.; Schlegelberger, B.; Leo, F.; Steinemann, D.; et al. CDKN2A and PIK3CA mutation in myoepithelial-like metaplastic breast cancer. Pathol.2018, 245, 373–383.
- Stradella, A.; Gargallo, P.; Cejuela, M.; Petit, A.; Bosch-Schips, J.; Carbonell, P.; Recalde, S.; Vethencourt, A.; Fernandez-Ortega, A.; Falo, C.; et al. Genomic characterization and tumor evolution in paired samples of metaplastic breast carcinoma. Modern Pathology 2022, in press.
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.; Van Loo, P.; Haughland, H.K.; Lilleng, P.K.; et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 2017, 32, 169–184.
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The genomic landscape of endocrine-resistant advanced breast cancer. Cancer Cell 2018, 34, 427–438.
- Bertucci, F.; Ng, C.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, V.; Kumal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564.
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Messard, C.; Levy, C.; Arnedos, M.; Lavroix-Triki, M.; et al. Mutational profile of metastatic breast cancers: A retrospective analysis. PLoS Med.2016, 13, e10002201.
- Paul, M.R.; Pan, T.; Pant, D.K.; Shih, N.; Chen, Y.; Harvey, K.L.; Solomon, A.; Lieberman, D.; Morrissette, J.; Soucier-Ernst, D.; et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J Clin Invest 2020, 130, 4252-4265.
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fildago, J.A.; Cristofanili, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res 2014, 20, 1757-1767.
- Zundelevich, A.; Dadiani, M.; Kahana-Edwin, S.; Itay, A.; Sella, T.; Gadot, M.; Cesarkas, K.; Farage-Barhom, S.; Saar, E.G.; Eyal, E.; et al. ESR1 mutations are frequent in newly diagnosed metastatic and loco-regional recurrence of endocrine-treated breast cancer and carry worse prognosis. Breast Cancer Res 2020, 22, 16.
- Jeselsohn, R.; Bergholz, J.S.; Pun, M.; Cornwell, M., Liu, W.; Nardone, A.; Xiao, T.; Li, W.; Xiu, X.; Buchwalter, G.; et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 2018, 33, 173-186.
- Li, Z.; Wu, Y.; Yates, M.E.; Tasdemir, N.; Bahreini, A.; Chen, J.; Levine, K.M.; Priedigkeit, N.M.; Nasrazadani, A.; Ali, S.; Buluwela, L.; et al. Hotspot ESR1 mutations are multimodal and contextual modulators of breast cancer metastasis. Cancer Res 2022, in press.
- Desmedt, C.; Pingitore, J.; Rothé, F.; Marchio, C.; Clatot, F.; Rouas, G.; Richard, F.; Bertucci, F.; Mariani, O.; Galant, C.; et al. ESR1 mutations in metastatic lobular breast cancer patients. NPJ Breast Cancer 2019, 5, 9.
- Bidard, F.; Callens, C.; Dalenc, F.; Pistilli, B.; De La Motte Rouge, T.; Clatot, F.; D’Hondt, V.; Teixeira, L.; Vegas, H.; Everhard, S.; et al. Prognostic impact of ESR1 mutations in ER+ HER2-MBC patients prior treated with first line AI and Palbociclib: an exploratory analysis of the PADA-1 trial. J Clin Oncol 2020, 38, suppl., abst. 1010.
- Bidard, F.C.; Hardy-Bessard, A.C.; Bachelot, T.; Pierga, J.Y.; Canon, J.L.; Clatot, F.; Andre, F.; De La Motte Rouge, T.; Pistilli, B.; Dalenc, F.; et al. Fulvestrant-palbociclib vs continuing aromatase inhibitor-palbociclib upon detection of circulating ESR1 mutation in HR+ HER2- metastatic breast cancer patients: results of PADA-1, a UCBG-GINECO randomized phase 3 trial. San Antonio Breast Cancer Symposium, 10-12, 2021, abst. GS3-05.
- Cristofanili, M.; DeMichele, A.; Giorgetti, C.; Turner, N.C.; Slamon, D.J.; Im, S.A.; Masuda, N.; Verma, S.; Loi, S.; Colleoni, M.; et al. Predictors of prolonged benefit from palbociclib plus fulvestrant in women with endocrine-resistant hormone receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer in PALOMA-3. Eur J Cancer 2018, 104, 21-31.
- Cristofanili, M.; Rugo, H.S.; Im, S.A.; Slamon, D.J.; Hrabeck, N.; Bondarenko, I. Overall survival (OS) with palbociclib (PAL) + fulvestrant (FUL) in women with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-) advanced breast cancer (ABC): updated analyses from PALOMA-3. J Clin Oncol 2021, 39, suppl.,abst. 1000.
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: new insights into molecular mechanisms of breast cancer metastasis. NPJ Precis Oncol 2018, 2, 4.
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cehang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K.; et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 28, 3271-3278.
- Cha, S.; Lee, E.; Wong, H.H. Comprehensive characterization of distinct genetic alterations in metastatic breast cancer across various metastatic sites. NPJ Breast Cancer 2021, 7, 93.
- Rinaldi, J.; Sokol, E.S.; Hartmaier, R.J.; Trabucco, S.E.; Grampton, G.M.; Goldberg, L.A.; Albacker, L.A.; Daemen, A. The genomic landscape of metastatic breast cancer: insights from 11,000 tumors. PLOS ONE 2020, 15, e0231999.
- Jiang, B.; Mu, Q.; Qiu, F.; Li, X.; Xu, W.; Yu, J.; Fu, W.; Cao, Y.; Wang, J. Machine learning of genomic features in organotropic metastases stratifies progression risk of primary tumors. Nat Commun 2021, 12, 6692.
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat Genet 2020, 52, 701-708.
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563-575.
- Tian, C.; Liu, S.; Wang, Y.; Song, X. Prognosis and genomic landscape of liver metastasis in patients with breast cancer. Front Oncol 2021, 11, 588136.
- Priedigkeit, N.; Hratmaier, R.J.; Chen, Y.; Vareslija, D.; Basudan, A.; Watters, R.J.; Thomas, R.; Leone, J.P.; Lucas, P.C.; Bhargava, R.; et al. Intrinsic subtype switching and acquired ERBB2/HER2 amplifications and mutations in breast cancer brain metastases. JAMA Oncol 2017, 3, 666-671.
- Vareslija, D.; Priedigkeit, N.; Fagan, A.; Purcell, S.; Cosgrove, N.; O’Halloran, P.J.; Ward, E.; Cocchiglia, S.; Hartmaier, R.; Castro, C.A.; et al. Transcriptome characterization of matched primary breast and brain metastatic tumors to detect novel actionable targets. J Natl Cancer Inst 2019, 111, djy110.
- Hulsbergen, A.F.; Claes, A.; Kavouridis, V.K.; Ansaripour, A.; Nagareda, C.; Highes, M.E.; Smith, T.R.; Brastianos, P.K.; Verhoeff, J.; Lin, N.U.; et al. Subtype switching in breast cancer brain metastases: a multicenter analysis. Neuro Oncol 2020, 22, 1173-1181.
- Cosgrove, N.; Vareslija, D.; Keelan, S.; Elangovan, A.; Atkinson, J.M.; Cocchiglia, S.; Bane, F.T.; Singh, V.; Furney, S.; Hu, C.; et al. Mapping molecular subtype specific alterations in breast cancer brain metastases identifies clinically relevant vulnerabilities. Nat Commun 2022, 13, 514.