Brain injury is a common cause of death and disability for people of all ages worldwide. Depending on the biomechanics, brain lesions may occur both in areas of the brain directly adjacent to the place of force application and in remote areas [4]. The mechanisms of hippocampal damage are of particular importance, since they underlie late complications of traumatic brain injury (TBI), such as epilepsy, depression and cognitive impairment. The mechanisms of reorganization of neuronal networks in the hippocampus include long-lasting chronic neuroinflammation and secondary damage to the nervous tissue. Responses and disturbances of the hypothalamic–pituitary–adrenal (HPA) axis may play a critical role in late post-traumatic pathology, in particular by modulation of synaptic activity and neuroinflammation in the hippocampus.
- brain trauma
- glucocorticoids
- corticosterone
- cortisol
- stress
- neuroinflammation
1. TBI, Its Late Consequences and the Hippocampus
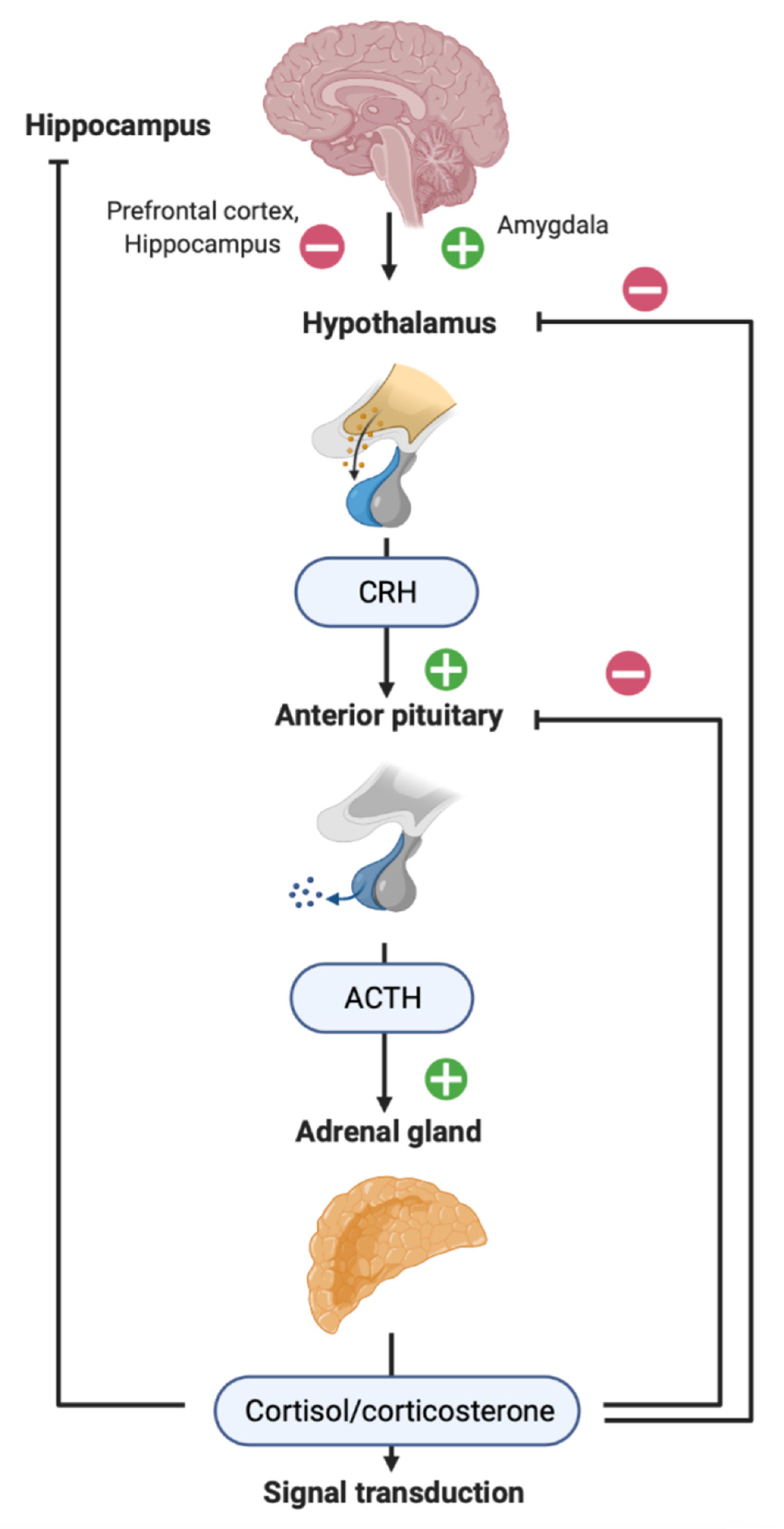
2. HPA Axis in Patients with TBI
3. Neuroinflammation and TBI
4. Neuroinflammation and GCs


This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10051139
References
- Annegers, J.F.; Hauser, W.A.; Coan, S.P.; Rocca, W.A. A population-based study of seizures after traumatic brain injuries. N. Engl. J. Med. 1998, 338, 20–24.
- Gupta, P.K.; Sayed, N.; Ding, K.; Agostini, M.A.; Van Ness, P.C.; Yablon, S.; Madden, C.; Mickey, B.; D’Ambrosio, R.; Diaz-Arrastia, R. Subtypes of Post-Traumatic Epilepsy: Clinical, Electrophysiological, and Imaging Features. J. Neurotrauma 2014, 31, 1439–1443.
- Malmgren, K.; Thom, M. Hippocampal sclerosis-Origins and imaging. Epilepsia 2012, 53, 19–33.
- Englander, J.; Bushnik, T.; Duong, T.T.; Cifu, D.X.; Zafonte, R.; Wright, J.; Hughes, R.; Bergman, W. Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Arch. Phys. Med. Rehabil. 2003, 84, 365–373.
- Haltiner, A.M.; Temkin, N.R.; Dikmen, S.S. Risk of seizure recurrence after the first late posttraumatic seizure. Arch. Phys. Med. Rehabil. 1997, 78, 835–840.
- Temkin, N.R. Risk Factors for Posttraumatic Seizures in Adults. Epilepsia 2003, 44, 18–20.
- Pohlmann-Eden, B.; Bruckmeir, J. Predictors and dynamics of posttraumatic epilepsy. Acta Neurol. Scand. 1997, 95, 257–262.
- Bombardier, C.H. Rates of Major Depressive Disorder and Clinical Outcomes Following Traumatic Brain Injury. JAMA 2010, 303, 1938.
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry 2019, 84, 1306–1328.
- Malykhin, N.V.; Carter, R.; Seres, P.; Coupland, N.J. Structural changes in the hippocampus in major depressive disorder: Contributions of disease and treatment. J. Psychiatry Neurosci. 2010, 35, 337–343.
- Hesdorffer, D.C.; Ishihara, L.; Mynepalli, L.; Webb, D.J.; Weil, J.; Hauser, W.A. Epilepsy, suicidality, and psychiatric disorders: A bidirectional association. Ann. Neurol. 2012, 72, 184–191.
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191.
- de Kloet, E.R.; Karst, H.; Joëls, M. Corticosteroid hormones in the central stress response: Quick-and-slow. Front. Neuroendocrinol. 2008, 29, 268–272.
- Maggio, N.; Segal, M. Corticosteroid Regulation of Synaptic Plasticity in the Hippocampus. Sci. World J. 2010, 10, 462–469.
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322.
- Kusmenkov, T.; Braunstein, M.; Schneider, H.; Bidlingmaier, M.; Prall, W.; Flatz, W.; Boecker, W.; Bogner, V. Initial free cortisol dynamics following blunt multiple trauma and traumatic brain injury: A clinical study. J. Int. Med. Res. 2019, 47, 1185–1194.
- Kakati, A.; Devi, B.I.; Bhadrinarayan, V.; Kalra, P.; Shukla, D. Endocrine dysfunction following traumatic brain injury in acute stage. Indian J. Neurotrauma 2013, 10, 92–96.
- Rao, T.P. A study of serum cortisol levels in acute head injury patients. J. Basic Clin. Physiol. Pharmacol. 2020, 32, 20190136.
- Agha, A.; Rogers, B.; Mylotte, D.; Taleb, F.; Tormey, W.; Phillips, J.; Thompson, C.J. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin. Endocrinol. 2004, 60, 584–591.
- Bensalah, M.; Donaldson, M.; Aribi, Y.; Iabassen, M.; Cherfi, L.; Nebbal, M.; Medjaher, M.; Haffaf, E.; Abdennebi, B.; Guenane, K.; et al. Cortisol evaluation during the acute phase of traumatic brain injury-A prospective study. Clin. Endocrinol. 2018, 88, 627–636.
- Bernard, F.; Outtrim, J.; Lynch, A.G.; Menon, D.K.; Matta, B.F. Hemodynamic Steroid Responsiveness is Predictive of Neurological Outcome After Traumatic Brain Injury. Neurocrit. Care 2006, 5, 176–179.
- Tanriverdi, F.; Schneider, H.J.; Aimaretti, G.; Masel, B.E.; Casanueva, F.F.; Kelestimur, F. Pituitary Dysfunction After Traumatic Brain Injury: A Clinical and Pathophysiological Approach. Endocr. Rev. 2015, 36, 305–342.
- Saichan, X.; Wei, C.; Qinglong, F.; Jun, W.; Lei, X. Plasma cortisol as a noninvasive biomarker to assess severity and prognosis of patients with craniocerebral injury. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3835–3838.
- Sörbo, A.; Eiving, I.; Theodorsson, E.; Rydenhag, B.; Jonsdottir, I.H. Pre-traumatic conditions can influence cortisol levels before and after a brain injury. Acta Neurol. Scand. 2020, 141, 342–350.
- Spikman, J.M.; van der Horn, H.J.; Scheenen, M.E.; de Koning, M.E.; Savas, M.; Langerak, T.; van Rossum, E.F.C.; van der Naalt, J. Coping with stress before and after mild traumatic brain injury: A pilot hair cortisol study. Brain Inj. 2021, 35, 871–879.
- Bay, E.; Sikorskii, A.; Gao, F. Functional Status, Chronic Stress, and Cortisol Response After Mild-to-Moderate Traumatic Brain Injury. Biol. Res. Nurs. 2009, 10, 213–225.
- Bay, E.; Hagerty, B.; Williams, R.A.; Kirsch, N. Chronic Stress, Salivary Cortisol Response, Interpersonal Relatedness, and Depression Among Community-Dwelling Survivors of Traumatic Brain Injury. J. Neurosci. Nurs. 2005, 37, 4–14.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153.
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting traumatic brain injury: From molecular mechanisms to therapeutic interventions. Biomedicines 2020, 8, 389.
- Vezzani, A.; Auvin, S.; Ravizza, T.; Aronica, E. Glia-Neuronal Interactions in Ictogenesis and Epileptogenesis: Role of Inflammatory Mediators. In Jasper’s Basic Mechanisms of the Epilepsies , 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M., Olsen, R., Delgado-Escueta, A., Eds.; Oxford University Press: Oxford, UK, 2012; ISBN 9780199746545.
- Vezzani, A.; Balosso, S.; Ravizza, T. Inflammation and epilepsy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 107, pp. 163–175. ISBN 9780444528988.
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208.
- Tobin, R.P.; Mukherjee, S.; Kain, J.M.; Rogers, S.K.; Henderson, S.K.; Motal, H.L.; Rogers, M.K.N.; Shapiro, L.A. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol. Commun. 2014, 2, 143.
- Hicks, R.; Soares, H.; Smith, D.; McIntosh, T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996, 91, 236–246.
- Cortez, S.C.; McIntosh, T.K.; Noble, L.J. Experimental fluid percussion brain injury: Vascular disruption and neuronal and glial alterations. Brain Res. 1989, 482, 271–282.
- Lescot, T.; Fulla-Oller, L.; Po, C.; Chen, X.R.; Puybasset, L.; Gillet, B.; Plotkine, M.; Meric, P.; Marchand-Leroux, C. Temporal and Regional Changes after Focal Traumatic Brain Injury. J. Neurotrauma 2010, 27, 85–94.
- Xu, S.; Sun, Q.; Fan, J.; Jiang, Y.; Yang, W.; Cui, Y.; Yu, Z.; Jiang, H.; Li, B. Role of Astrocytes in Post-traumatic Epilepsy. Front. Neurol. 2019, 10, 1149.
- Thrane, A.S.; Rangroo Thrane, V.; Nedergaard, M. Drowning stars: Reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014, 37, 620–628.
- Tapp, Z.M.; Godbout, J.P.; Kokiko-Cochran, O.N. A Tilted Axis: Maladaptive Inflammation and HPA Axis Dysfunction Contribute to Consequences of TBI. Front. Neurol. 2019, 10, 345.
- D’amico, R.; Salinaro, A.T.; Fusco, R.; Cordaro, M.; Impellizzeri, D.; Scuto, M.; Ontario, M.L.; Dico, G.L.; Cuzzocrea, S.; Di Paola, R.; et al. Hericium erinaceus and coriolus versicolor modulate molecular and biochemical changes after traumatic brain injury. Antioxidants 2021, 10, 898.
- Chiu, C.-C.; Liao, Y.-E.; Yang, L.-Y.; Wang, J.-Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.-Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49.
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167.
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247.
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33–39.
- Frank, M.G.; Miguel, Z.D.; Watkins, L.R.; Maier, S.F. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain. Behav. Immun. 2010, 24, 19–30.
- Espinosa-Oliva, A.M.; de Pablos, R.M.; Villarán, R.F.; Argüelles, S.; Venero, J.L.; Machado, A.; Cano, J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol. Aging 2011, 32, 85–102.
- Munhoz, C.D. Chronic Unpredictable Stress Exacerbates Lipopolysaccharide-Induced Activation of Nuclear Factor- B in the Frontal Cortex and Hippocampus via Glucocorticoid Secretion. J. Neurosci. 2006, 26, 3813–3820.
- Munhoz, C.D.; Sorrells, S.F.; Caso, J.R.; Scavone, C.; Sapolsky, R.M. Glucocorticoids Exacerbate Lipopolysaccharide-Induced Signaling in the Frontal Cortex and Hippocampus in a Dose-Dependent Manner. J. Neurosci. 2010, 30, 13690–13698.
- Komoltsev, I.G.; Tret’yakova, L.V.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Butuzov, A.V.; Novikova, M.R.; Kvichansky, A.A.; Moiseeva, Y.V.; Onufriev, M.V.; et al. Neuroinflammatory Cytokine Response, Neuronal Death, and Microglial Proliferation in the Hippocampus of Rats During the Early Period After Lateral Fluid Percussion-Induced Traumatic Injury of the Neocortex. Mol. Neurobiol. 2021, 59, 1151–1167.
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int. J. Mol. Sci. 2021, 22, 5883.
- Tretyakova, L.V.; Kvichansky, A.A.; Bolshakov, A.P.; Gulyaeva, N.V. Dexamethasone Modulates Lipopolysaccharide-Induced Expression of Proinflammatory Cytokines in Rat Hippocampus. Neurochem. J. 2021, 15, 302–307.
- Skupio, U.; Tertil, M.; Sikora, M.; Golda, S.; Wawrzczak-Bargiela, A.; Przewlocki, R. Behavioral and molecular alterations in mice resulting from chronic treatment with dexamethasone: Relevance to depression. Neuroscience 2015, 286, 141–150.
- Frank, M.G.; Hershman, S.A.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 2014, 40, 191–200.
- Fenn, A.M.; Gensel, J.C.; Huang, Y.; Popovich, P.G.; Lifshitz, J.; Godbout, J.P. Immune Activation Promotes Depression 1 Month After Diffuse Brain Injury: A Role for Primed Microglia. Biol. Psychiatry 2014, 76, 575–584.
- Dong, T.; Zhi, L.; Bhayana, B.; Wu, M.X. Cortisol-induced immune suppression by a blockade of lymphocyte egress in traumatic brain injury. J. Neuroinflamm. 2016, 13, 197.
- Raefsky, S.M.; Mattson, M.P. Adaptive responses of neuronal mitochondria to bioenergetic challenges: Roles in neuroplasticity and disease resistance. Free Radic. Biol. Med. 2017, 102, 203–216.