After the identification of a biological target (enzyme, receptor, protein and so on), the focus of the early phase of drug discovery rests on the identification of leads or compounds that exhibit pharmacological activity against this specific target. Compounds of interest are most often discovered in pre-existing libraries of compounds that can be either virtual or physical. Computer-aided methods which have become increasingly important over the years in drug development utilize virtual compound libraries. While physical compound libraries reach the number of millions of molecules, virtual compound libraries created by large pharmaceutical companies can range from 107 to 1018 molecules. Investigations of these libraries identifies specific molecules, synthetic pathways and focus on a specific chemical space. Targeted libraries are often smaller and are focused towards a specific chemical space. They are created by using relevant biological information with the aim to decrease the processing time associated with larger libraries while maintaining the most relevant chemical space where lead compounds can be found. Due to the fact that they required less computational or wet-lab labor to process they have become very popular with smaller laboratories which try to compete in the drug-development sector. Many modern vendors of compounds today offer such libraries, but the quality of the procedure used to define desired chemical space and select compounds is questionable.
- targeted libraries
- focused libraries
- computer-aided drug design
1. The role of Targeted Libraries
As seen in (Figure1) the compound library plays a crucial role in the process from target identification to drug candidate. the computer-aided drug design (CADD) methodology is nowadays routinely used in drug discovery, the need for virtual compound libraries increases. Consequently, numerous commercial vendors have emerged, offering such libraries in both physical and virtual forms. To further facilitate the drug discovery, commercial vendors are now offering scientists the so-called targeted or focused compound libraries ready to jumpstart the drug design and discovery in a specific field. These focused libraries often represent a subset of compounds from the manufacturer’s full database that may possess specific properties for a selected target or drug design field [1].
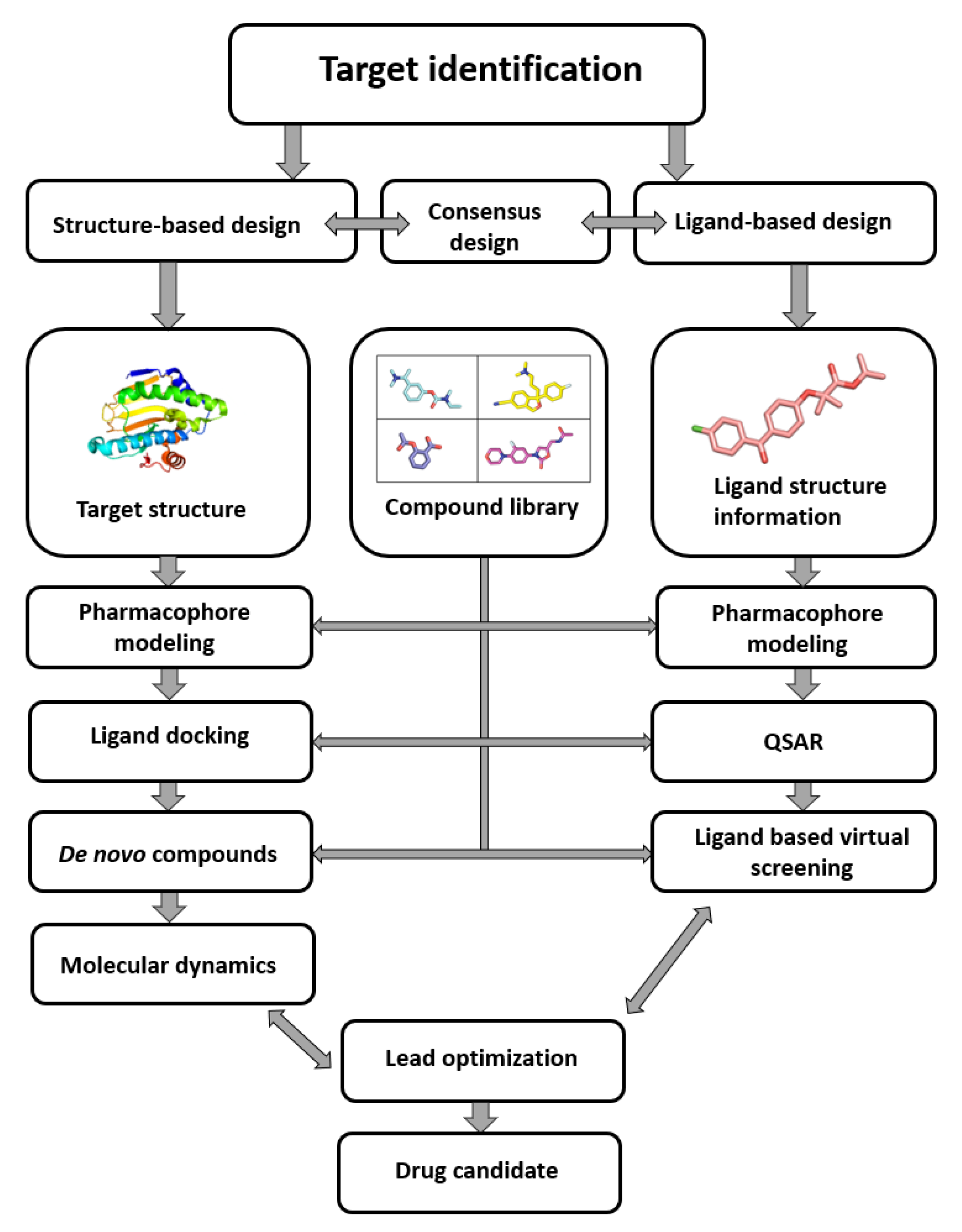
Figure 1. Computer-aided drug design process chart used to obtain lead compounds [2].
2. Preparation of Targeted Libraries
When designing or examining an existing molecular library, a researcher should be aware of important steps and drawbacks associated with virtual molecules. After obtaining the target structure and learning about the molecular and biological context, a researcher can proceed toward designing the virtual compound library.
The main goal of targeted libraries is to cover a diverse chemical space with as few compounds as possible. Due to the fast-growing availability of the chemical and biological structural data, the public bioactivity databases provide an excellent starting point [3]. The most comprehensive and curated information about molecules is freely available in the prominent PubChem, ChEMBL and ZINC databases. After assembling the initial database from various sources, the next step is to remove duplicates, so that only unique structures are processed further [3]. The choice of the chemical file format is vital, as it dictates how the obtained data can be used. The most recommended file formats for representing molecules as strings include the SMILES and InChI formats. In most situations, multiple SMILES strings can equally well represent a single molecule. The application of the canonical SMILES, which uses only a single string per molecule, is recommended to avoid duplication and filtering problems. For the spatial representation of molecules, either the Structure Data Format (SDF) or the MDL Molfile (MOL) format are the most common [4]. Online libraries can usually be downloaded in these formats, making it easier to obtain a coherent library. When performing filtering, clustering or similarity searches, the SMILES format is preferred, as it leads to faster processing, due to its string representation; however, the spatial information is required for downstream methods such as 3D pharmacophore development, molecular docking and molecular dynamics. The molecular representation always requires the extra care and exploration in terms of conformational viability, chiral centers, tautomerism, compound ionization, presence of salts, structural faults and so on By default, the hydrogen atoms are often not present in various chemical file formats and should be added during the library preparation. Tautomerism represents a property of chemical compounds that affects the calculation of their physiochemical properties, such as logP, logD and pKa, and subsequently bears consequences in both QSAR and molecular docking [5]. Due to their different structural representations, tautomers are often handled as separate molecules by CADD programs [3][6]. Moreover, since proteins are known to be enantioselective toward the binding ligands, exploring chirality when designing a virtual library is an important aspect to consider. In the majority of virtual libraries, however, compounds are represented by a single stereoisomer, or the stereo information is absent altogether. Exploring chirality thoroughly will expand the database size by 2n per molecule, where n is the number of chiral centers present. With larger databases, this issue will be even more pronounced and should be considered before generating all possible stereoisomers [7]. In general, it is recommended to explore unspecified chirality, which should be performed on a case-by-case basis with regard to the biological context [3]. Furthermore, for compounds that have ionizable groups, multiple different structural representations should be used. Within a reasonable pH range, structures should be represented as protonated and deprotonated forms of compounds [7]. The biological context of the target protein should be used to provide an accurate representation of the environment. After the final compound 3D structure generation, energy minimization should be carried out in order to optimize the molecular geometry [8]. Library design should factor in the avoidance of toxic outcomes. Despite the fact that toxicity of drugs is multifactorial and that predicting the exact property responsible for toxicity is difficult, several correlations of toxicity to in vitro pharmacology profiles exist and can be translated to in silico tools which examine molecular descriptors and filter the libraries accordingly [9]. The filtering itself can also flag compounds with reactive functional groups or moieties, optically interfering components, aggregators or frequent hitters. The filtering of “unwanted” molecular species using computational filters represents an essential element of library preparation which should always be considered in a suitable context [10][11]. The general guide for virtual compound library preparation is presented in (Figure 2).
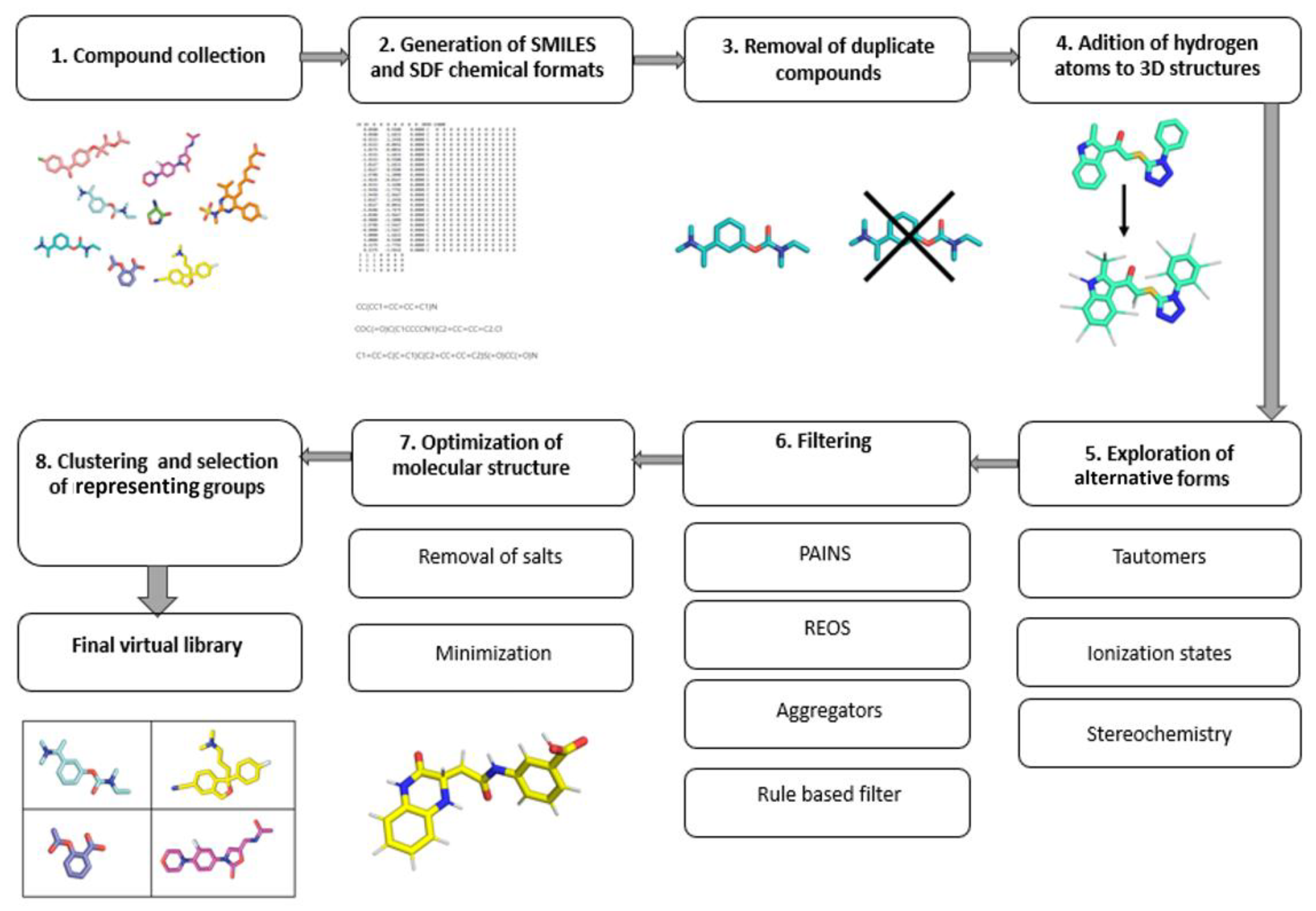
Figure 2. Workflow of an efficient library preparation for medicinal chemistry.
3. Examples of Commercial Targeted Libraries
3.1. Enamine
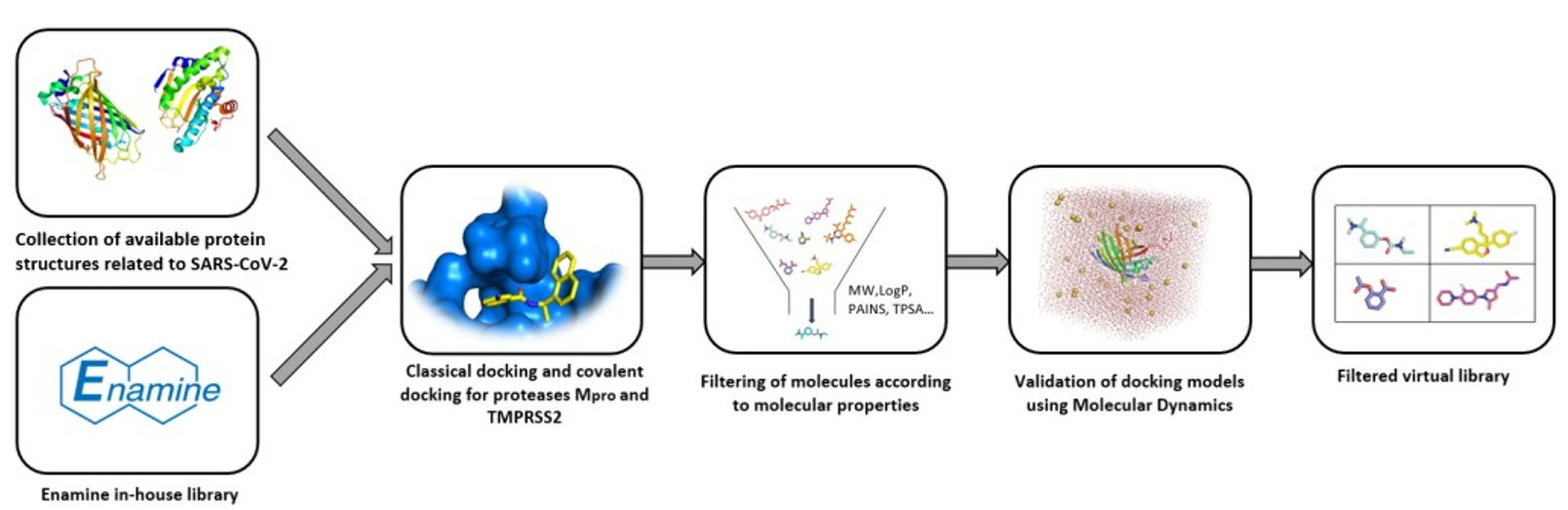
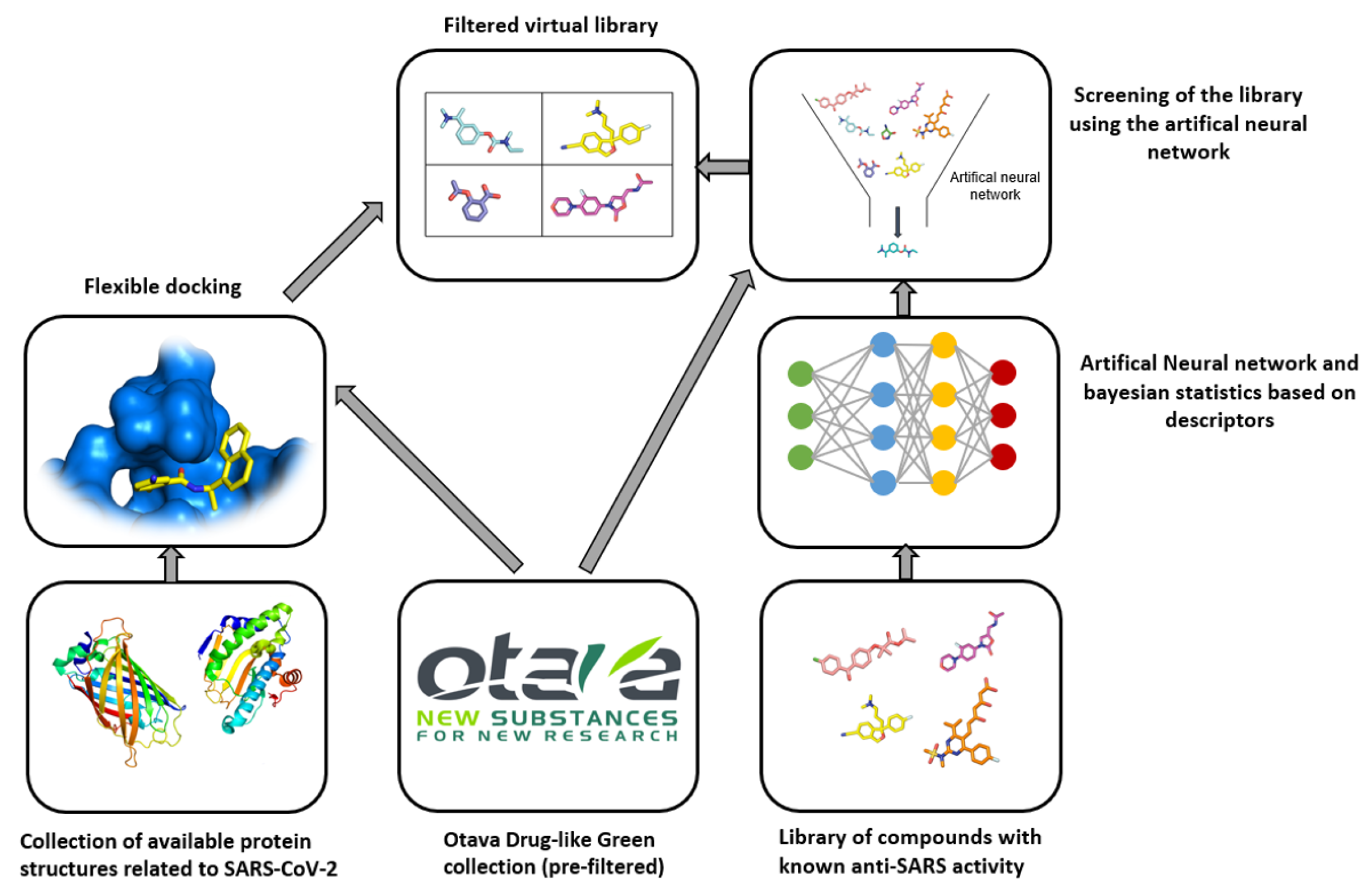
3.2. Otava
3.3. Life Chemicals
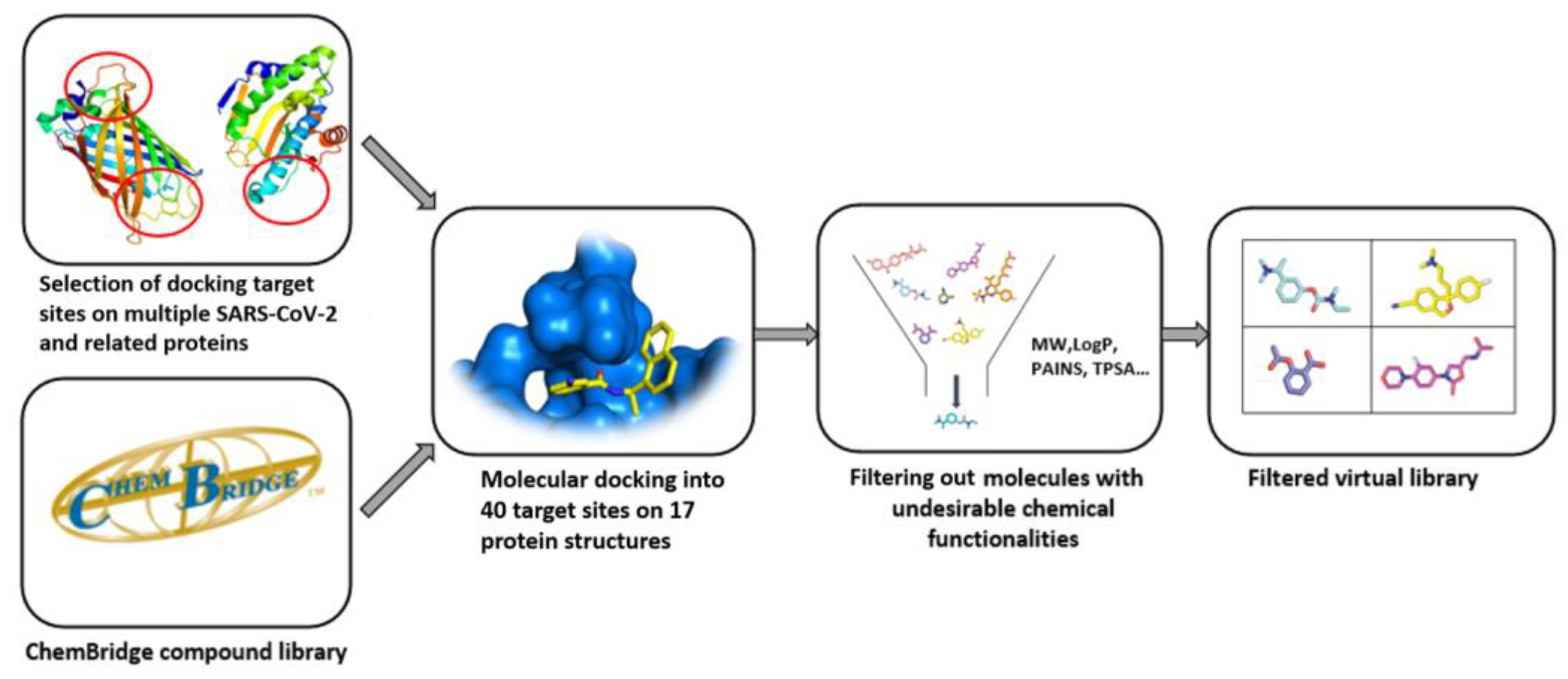
4. Quality of Commercial Targeted Libraries
The main problem with the libraries discussed, in their opinion, is that vendors lack the information on library design, as well as the references to the primary literature. Commercial vendors consistently provide only general information about the screening protocols used to design the target libraries offered and are even less informative about the actual filters used. No references to actual actives are provided when using ligand-based design, with the exception of marketed libraries of known actives that can be referenced post-purchase by the client through databases such as ChEMBL. For structure-based approaches, usually only target or protein classes or a general panel of targets are provided, with reference to methods such as molecular docking. No precise docked receptors or PDB IDs of the targets are available, and no docking protocols or even references to the molecular docking software (or other HTVS software) are provided. This fact is that worrying does not bode well for the use of these libraries in drug design and lends itself to commercial drug companies to focus on and improve. In the more than 10 years of experience, it has been found that the quality of the compounds purchased from commercial suppliers is usually high, with most compounds being characterized by NMR and MS/HRMS analyses after purchase, and their purity being determined to be about 95% or higher by HPLC. However, analytical data are not part of the original catalogue selection, and purity data are not usually available prior to the purchase. Therefore, it was recommend that the reader be aware of this fact and even take advantage of asking the commercial vendor for characterization and purity data prior to the purchase (in some situations, NMR, MS and HPLC data can be obtained). Nowadays, vendors can even point out certain availability and quality issues. From the time of purchase, the quality of the supply chain (cold storage, if necessary, and insurance for possible shipping problems) and the emphasis on quality storage after delivery are essential [12].
5. Summary
Since December 2019, the new SARS-CoV-2-related COVID-19 disease has caused a global pandemic and shut down the public life worldwide. Several proteins have emerged as potential therapeutic targets for drug development, and we sought out to review the commercially available and marketed SARS-CoV-2-targeted libraries ready for high-throughput virtual screening (HTVS). We evaluated the SARS-CoV-2-targeted, protease-inhibitor-focused and protein-protein-interaction-inhibitor-focused libraries to gain a better understanding of how these libraries were designed. The most common were ligand- and structure-based approaches, along with various filtering steps, using molecular descriptors. Often, these methods were combined to obtain the final library. We recognized the abundance of targeted libraries offered and complimented by the inclusion of analytical data; however, serious concerns had to be raised. Namely, vendors lack the information on the library design and the references to the primary literature. Few references to active compounds were also provided when using the ligand-based design and usually only protein classes or a general panel of targets were listed, along with a general reference to the methods, such as molecular docking for the structure-based design. No receptor data, docking protocols or even references to the applied molecular docking software (or other HTVS software), and no pharmacophore or filter design details were given. No detailed functional group or chemical space analyses were reported, and no specific orientation of the libraries toward the design of covalent or noncovalent inhibitors could be observed. All libraries contained pan-assay interference compounds (PAINS), rapid elimination of swill compounds (REOS) and aggregators, as well as focused on the drug-like model, with the majority of compounds possessing their molecular mass around 500 g/mol. These facts do not bode well for the use of the reviewed libraries in drug design and lend themselves to commercial drug companies to focus on and improve.
For further details the reader should reference the article by Kralj, Jukič and Bren: Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein-Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design[13].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23010393
References
- Paul H. Bernardo; Joo Chuan Tong; In Silico Design of Small Molecules. Springer Protocols Handbooks 2011, 800, 25-31, 10.1007/978-1-61779-349-3_3.
- Gregory Sliwoski; SandeepKumar Kothiwale; Jens Meiler; Edward W. Lowe Jr.; Computational Methods in Drug Discovery. Pharmacological Reviews 2013, 66, 334-395, 10.1124/pr.112.007336.
- Cristian G. Bologa And Tudor I. Oprea Marius M. Olah; Cristian Bologa; Tudor Oprea; Strategies for Compound Selection. Current Drug Discovery Technologies 2004, 1, 211-220, 10.2174/1570163043334965.
- Arthur Dalby; James G. Nourse; W. Douglas Hounshell; Ann K. I. Gushurst; David L. Grier; Burton A. Leland; John Laufer; Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. Journal of Chemical Information and Computer Sciences 1992, 32, 244-255, 10.1021/ci00007a012.
- Yvonne Connolly Martin; Let’s not forget tautomers. Journal of Computer-Aided Molecular Design 2009, 23, 693-704, 10.1007/s10822-009-9303-2.
- Frank Oellien; Joerg Cramer; Carsten Beyer; Wolf-Dietrich Ihlenfeldt; Paul M. Selzer; The Impact of Tautomer Forms on Pharmacophore-Based Virtual Screening.. ChemInform 2007, 38, 2342-2345, 10.1002/chin.200707206.
- W. C. Guida; K. G. Daniel; The Significance of Chirality in Drug Design and Development. Current Topics in Medicinal Chemistry 2011, 11, 760-770, 10.2174/156802611795165098.
- Elaine C. Meng; Daniel A. Gschwend; Jeffrey M. Blaney; Irwin D. Kuntz; Orientational sampling and rigid-body minimization in molecular docking. Proteins: Structure, Function, and Bioinformatics 1993, 17, 266-278, 10.1002/prot.340170305.
- David A Price; Julian Blagg; Lyn Jones; Nigel Greene; Travis Wager; Physicochemical drug properties associated within vivotoxicological outcomes: a review. Expert Opinion on Drug Metabolism & Toxicology 2009, 5, 921-931, 10.1517/17425250903042318.
- Elena Lenci; Andrea Trabocchi; Peptidomimetic toolbox for drug discovery. Chemical Society Reviews 2020, 49, 3262-3277, 10.1039/d0cs00102c.
- Tian Zhu; Shuyi Cao; Pin-Chih Su; Ram Patel; Darshan Shah; Heta B. Chokshi; Richard Szukala; Michael E. Johnson; Kirk E. Hevener; Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis. Journal of Medicinal Chemistry 2013, 56, 6560-6572, 10.1021/jm301916b.
- Sebastjan Kralj; Marko Jukič; Urban Bren; Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. International Journal of Molecular Sciences 2021, 23, 393, 10.3390/ijms23010393.
- Sebastjan Kralj; Marko Jukič; Urban Bren; Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. International Journal of Molecular Sciences 2021, 23, 393, 10.3390/ijms23010393.