Completely randomizing even relatively short peptides would require a library size surpassing the capacities of most platforms. Sampling the complete mutational space for peptides exceeding 8–9 residues is therefore practically impossible, and gene diversification strategies only allow for generation and subsequent interrogation of a limited subset of the entire theoretical peptide population. Directed evolution of peptides therefore strives to ascend towards peak activity through mutational steps, accumulating beneficial mutational over several generations, resulting in improved phenotype. We briefly discuss combinatorial library platforms and take an in-depth look at diversification techniques for random and focused mutagenesis.
- combinatorial peptide library
- random mutagenesis
- focused mutagenesis
Introduction
A combinatorial peptide library is a collection of fully or partially randomized peptides of defined length. Peptide libraries are classified either as synthetic (non-biological) or biological. As the name implies, synthetic libraries are produced through combinatorial peptide synthesis by allowing coupling of more than one amino acid residue per position. Here, individual peptides can be coupled to (and in fact synthesized on) beads in a one-bead-one-compound setting [1] and screened against a fluorophore-labelled target using fluorescence-activated sorting (Fig. 1A). An alternative approach to synthetic library screening is positional scanning, where the peptide library is divided into sub-libraries, each having one of the positions defined (i.e., occupied by a specific amino acid residue), while the rest of the peptide structure is randomized (Fig. 1B). Each of the sub-libraries is assayed for activity to deduce the consensus peptide sequence with optimal activity [2]. Conversely, in iterative deconvolution strategy optimal residues at individual positions are identified in a step-by-step fashion (Fig. 1C). Sub-libraries of randomized peptides but with different defined residues at first position are screened for activity, and the optimal residue in that position is retained in the second generation sub-library which contains peptides with different defined residues at the second position. This approach is repeated until all the positions have been interrogated [2]. The term biological library signifies that the peptides are produced on ribosomes. Here, it is essential that the phenotype (the encoded peptide) and the genotype (the encoding nucleic acid sequence) are physically linked, allowing correlation of peptide sequence and genetic information during screening, and identification of hits via nucleic acid sequencing [3]. In library platforms such as bacterial and yeast display, the peptides are expressed from fusions genes in frame with a gene for a cell surface protein. In phage display, peptides are anchored to viral protein coat, encapsulating genetic information, through fusions with capsid structural proteins. In ribosome and mRNA display, peptides are coupled to mRNA indirectly (non-covalently via ribosome complex) or directly (covalently via puromycin), respectively.
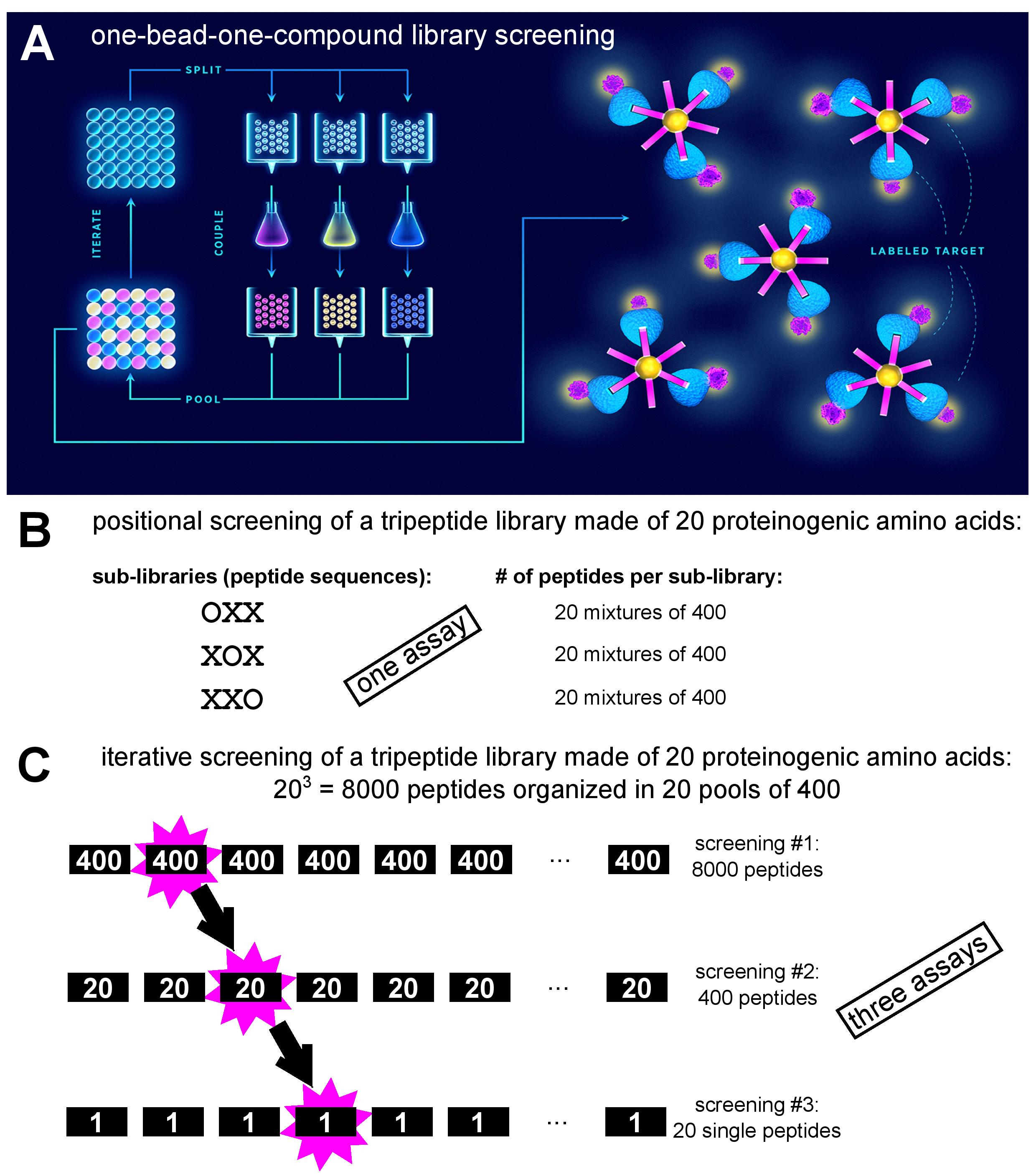
Figure 1: Depiction of various synthetic peptide library screening approaches. A. One-bead-one-compound library synthesis by the ‘split-and-mix’ method and screening via fluorescence-activated bead sorting after incubation with fluorophore-labelled target (adapted from [4]). B. Example of positional screening of a tripeptide library: 60 peptide mixtures are screened in parallel in a single assay (adapted from [2]). C. Example of iterative screening of a tripeptide library: three consecutive screens are performed with sub-libraries of decreasing diversity (adapted from [2]).
In the following sections, diversification and construction of biological combinatorial peptide libraries are reviewed. As the peptides are genetically encoded, the diversification step is performed at the level of DNA, either by synthesis or mutagenesis.
Peptide Library Design and Construction
Peptide maturation can be depicted as an ascent in a simplified fitness landscape (Fig. 2) in which the x-y coordinates denote the otherwise multidimensional genotype, and the z-axis represent the peptide’s “phenotypic” traits, e.g., target affinity. Ascending towards peak activity with mutational steps is the goal of directed evolution. Beneficial mutations accumulate over several generations upon selection pressure, resulting in improved phenotype [5].

Figure 2: Maturation of a peptide depicted as ascent on a simplified fitness landscape. After each selection round, mutations are introduced into the enriched combinatorial library, and the next generation of peptides is screened for improved affinity and/or activity (adopted from [4]).
In general, library generation can be performed either through focused or random mutagenesis. The latter is usually used in the absence of structure-function relationship knowledge. In focused mutagenesis residues previously found to be essential for peptide activity are retained (or favoured over the rest of the building block set), while the others are (fully or partially) randomized. Of course, the odds that a library contains improved peptide variants are higher for those produced by focused mutagenesis. A plethora of mutagenesis methods can be used for gene diversification in library generation and we will briefly discuss them below.
Random Mutagenesis
Random mutagenesis based on physical and/or chemical mutagens is sufficient for traditional genome screening (gene inactivation), but it is not suitable for directed evolution due to limited mutational spectrum [6,7]. For library generation purposes, random mutagenesis can be performed in vivo in bacterial mutator strains that contain defective proofreading and repair enzymes (mutS, mutT, and mutD) [8–10]. Another approach in E. coli relies on mutagenesis plasmids (MP), which carry multiple genes for proteins affecting DNA proofreading, mismatch repair, translesion synthesis, base selection, and base excision repair, thereby enabling broad mutagenic spectra. MPs support mutation rates 322,000-fold over basal levels and are suitable for platforms based on bacterial and phage-mediated directed evolution [11]. Unfortunately, beside the library gene, mutator strains and MPs also induce deleterious mutations in host genome. In eukaryotes this was overcome by the development of orthogonal in vivo DNA replication apparatus, which in essence utilizes plasmid-polymerase pairs, limiting mutagenesis to a cytoplasm-only event [12]. Related phenomena are also known to occur in nature (e.g., the Bordetella bronchiseptica bacteriophage error-prone retroelement, which selectively introduces mutations into the gene encoding the major tropism determinant (Mtd) protein on the phage tail fibers [13]) and can be exploited for creating libraries [14].
One of the most established methods for in vitro random mutagenesis is the error-prone PCR (epPCR), first described in 1989 [15]. It works by harnessing the natural error rate of low-fidelity DNA polymerases, generating point mutations during PCR amplification. However, even the faulty Taq DNA polymerase is not erroneous enough to be useful for constructing combinatorial libraries under standard amplification conditions. The fidelity of the reaction can be further reduced by altering the amount of bivalent cations Mn2+ and Mg2+, introduction of biased concentrations of deoxyribonucleoside triphosphates (dNTPs) [16], using mutagenic dNTP analogues [17], or adjusting elongation time and the number of cycles [18]. Random mutations can also be induced by utilizing 3’-5’ proofreading-deficient polymerases [19–21].
Despite its popularity, epPCR suffers from limited mutational spectrum as it inclines to transitions (A↔G or T↔C). Thus, epPCR-generated libraries are abundant in synonymous and conservative nonsynonymous mutations as a result of codon redundancy [5]. Ideally, all four transitions (AT→GC and GC→AT) and eight transversions (AT→TA, AT→CG, GC→CG, and GC→TA) would occur at equal ratios, with the desired probability, and without insertions or deletions [22]. This problem has been addressed by the sequence saturation mutagenesis (SeSaM) [23] method, which utilizes deoxyinosine, a promiscuous base-pairing nucleotide that is enzymatically inserted throughout the target gene and later changed for canonical nucleotides using standard PCR amplification of the mutated template gene. SeSaM was later improved with the introduction of SeSaM-Tv-II [24], which generates sequence space unobtainable via conventional epPCR by increasing the number of transversions. It employs a novel polymerase with increased processivity, alowing efficient read through consecutive base-pair mismatches. EpPCR has been successfully adopted for library generation in various platforms, including phage, [25], E. coli [26] and ribosome [27] display.
Alternatively, mutagenesis can be achieved by performing isothermal rolling circle amplification (RCA) under error-prone conditions. Using a wild-type sequence as a template, this method is able to generate a random DNA mutant library, which can be directly transformed into E. coli without subcloning [28]. RCA was advanced further, coupling it with Kunkel mutagenesis [29] (see below). Termed ‘selective RCA’ (sRCA), it operates by producing plasmids in ung- (uracil-DNA-glycosylase deficient) dut- (dUTP diphosphatase deficient) E. coli strain to introduce non-specific uridylation (dT→dU). After PCR with mutagenic primers, abasic sites are created by the uracil-DNA glycosylase in the uracil-containing template. Only mutagenized products are amplified by RCA, excluding non-mutated background sequences [30].
Although epPCR generates high mutational rates, the sequence space remains mostly untapped [31]. DNA shuffling is touted to be superior to epPCR and oligonucleotide-directed mutagenesis because it does not suffer from the possibility of introducing neutral or non-essential mutations from repeated rounds of mutagenesis [32]. DNA shuffling was the first in vitro recombination method and it involves random fragmentation of a pool of closely related dsDNA sequences and subsequent reassembly of fragments by PCR [33]. Such template switching generates a myriad of new sequences and improves library diversity by mimicking natural sexual recombination [34]. Meyer et al. [31] developed an approach where DNase I creates double-stranded breaks at the regions of interest, followed by denaturation and reannealing at homologous regions. Hybridized fragments then serve as templates and are subjected to repeated PCR rounds to form a whole array of new sequences. Improved methods were developed, eliminating the lengthy DNA fragmentation step. In ‘staggered extension process’ (StEP), polynucleotide sequences can be diversified through severely-abbreviated annealing/polymerase-catalyzed extension. In each cycle, growing fragments switch between different templates and anneal to them based on sequence complementarity. They then extend further and the cycle is repeated until full-length mosaic sequences are formed [35]. Another ingenious method for creating random customized peptide libraries by Fujishima et al. [36] works by shuffling short DNA blocks with dinucleotide overhangs, enabling efficient and seamless library assembly through a simple ligation process.
Currently, recombination methods are shifting from in vitro to in vivo. Taking advantage of the high occurrence of homologous DNA recombination events in S. cerevisiae, ‘mutagenic organized recombination process by homologous in vivo grouping’ (MORPHING) method was developed. MORPHING is a ‘one-pot’ random mutagenesis method allowing construction of libraries with various degrees of diversity. Short DNA segments are produced by epPCR, and subsequently assembled with conserved overlapping gene fragments and the linearized plasmid by in vivo recombination upon transformation into yeast cells [37]. Another technique for assembling linear DNA fragments with homologous ends in E. coli is called ‘in vivo assembly’ (IVA). IVA uses PCR amplification with primers designed to substitute, delete or insert portions of DNA, and to simultaneously append homologous sequences at amplicon ends. Finally, it exploits recA-independent homologous recombination in vivo, greatly simplifying complex cloning operations. Thus, multiple simultaneous modifications (insertions, deletions, point mutations and/or site-saturation mutagenesis) are confined to a single PCR reaction, and multi-fragment assembly (library construction) proceeds in bacteria following transformation [38].
Focused Mutagenesis
Effectively exploring the sequence landscape requires structural and biochemical data (from previous random mutagenesis studies), which can be leveraged to constrain genetic variation to distinct positions of the (poly)peptide, such as regions of the peptide aptamer scaffold which can endure substitutions/insertions/deletions without affecting their overall protein fold, or those peptide residues considered not absolutely essential for specific property of interest (and whose mutation might further augment peptide’s activity). Random mutagenesis results in stochastic point mutations at codons corresponding to such residues, but systematically interrogating the entire set of residues at a specific position requires a focused mutagenesis strategy. Focused libraries are typically smaller and more effective, as they only address the residues presumed to bestow the peptide with the property of interest [39].
Enzyme-Based Approaches
Building a library of recombinant DNA constructs is a widely adopted practice accessible to virtually all laboratories, due to the ease of oligonucleotide synthesis and availability of commercial restriction enzymes and DNA ligases. The so-called oligonucleotide-directed mutagenesis enables point or multiple mutations to the target DNA sequence [40]. Normally, a mutagenic primer is designed and synthesized, subsequently elongated by Klenow fragment of DNA polymerase I, ligated into a vector by T4 DNA ligase and finally transformed into a competent E. coli strain. This process is long and includes multiple subcloning and ssDNA rescuing steps [41]. Several kits for site-specific mutagenesis based on mutagenic primers are commercially available. One of the systems works by applying a pair of forward and reverse complementary oligonucleotides with designed mutations. The primers are perfectly complementary to the template at 5’ and 3’ ends, but carry a changed central nucleotide sequence. A high-fidelity Pfu DNA polymerase is used to amplify the entire plasmid harboring the gene to be mutated, followed by the removal of the template by DpnI (an endonuclease specific for methylated DNA) [42]. There are numerous adaptations of this method (reviewed by Tee and Wong [22]).
An approach termed ‘Kunkel mutagenesis’ is commonly used for constructing libraries displayed on filamentous phage [43,44], because its genome is circular and single-stranded. In Kunkel mutagenesis, mutations are introduced with a mutagenic primer that is complementary to the circular ssDNA template. The template is propagated in an ung- dut- E. coli strain. This enzyme handicap results in the template DNA containing uracil bases in place of thymine. The template is recovered and hybridized with the primer and extended by polymerase, followed by transformation into ung+ dut+ host cells [29]. Upon transformation, uridylated DNA template is biologically inactivated through the action of uracil glycosylase [45] of the ung+ dut+ host, granting a strong selection advantage to the mutated strand(s) over the template.
Overlap extension PCR is another focused mutagenesis approach. First, two DNA fragments with homologous ends (and harboring desired mutation(s)) are amplified in separate PCR reactions by using 5’ complementary oligonucleotides. In a subsequent reaction, the fragments are combined; now, the overlapping 3’ ends from one of the strands of each fragment anneal and serve as ‘mega’ primers for extension of the complementary strands. Finally, the construct is amplified with the two flanking primers [46]. Based on this strategy, the SLIM (Site-directed Ligase-Independent Mutagenesis) method, compatible with all three types of sequence modifications (insertion, deletion, and substitution), employs an inverse PCR amplification of the plasmid-embedded template by two 5’ adapter-tailed long forward and reverse primers (which include modifications) and two short forward and reverse primers (identical to the long ones but lacking the 5’ adapter sequences) in a single reaction, producing 4 distinct amplicons. Next, the amplicons are heat denatured and reannealed to yield 16 (hetero)duplexes, 4 of which are directly cloneable, forming circular DNA through ligation-independent pathway via complementary 5’ and 3’ single-stranded overhangs. All steps of the SLIM procedure are carried out in a single tube [47].
Gibson assembly is a method of combining up to 15 DNA fragments containing 20-40 bp overlaps in a single isothermal reaction. It utilizes a cocktail of three enzymes; exonuclease, DNA polymerase, and DNA ligase. The exonuclease nibbles back DNA form the 5’ end, enabling annealing of homologous DNA fragments. DNA polymerase then fills in the gaps, followed by the covalent fragment joining by the DNA ligase [48]. Applications of Gibson Assembly include site-directed mutagenesis and library construction [49]. A recent adaptation, ‘QuickLib’, is a modified Gibson assembly method that has been used to generate a cyclic peptide library [50]. QuickLib uses two primers that share complementary 5’ ends; one long partially degenerate, and the other short non-degenerate, which are then used for full plasmid PCR amplification. Subsequently, a Gibson reaction is performed which circularizes the library of linear plasmids, followed by template elimination by DpnI restriction.
Besides conventional enzymes involved in cumbersome digestion and ligation steps, other enzymes can be utilized for mutagenesis. In nature, lambda exonuclease aids viral DNA recombination. It progressively degrades the 5′-phosphoryl strand of a duplex DNA from 5’ to 3’, producing ssDNA and (mono)nucleotides [51]. To exploit this property, first, a PCR amplification using template ssDNA and phosphorylated primers with overlapping regions is performed. The PCR product is then treated with lambda exonuclease, generating ssDNA fragments that are subsequently annealed via overlap regions. Afterwards, Klenow fragment is employed to create dsDNA. In this manner, site-specific mutagenesis can be performed using primers that contain degenerate bases [52].
One of the most broadly used approaches for characterization of individual amino acid residues of a (poly)peptide with regards to their contribution to binding affinity or activity is the alanine-scanning mutagenesis. As the name implies, the technique is based on systematic substitution of residues with alanine, and assessing ligand’s activity in a biochemical assay. Alanine eliminates the influence of all side chain atoms beyond the beta-carbon, thus exploring the role of side chain functional groups at interrogated positions [53]. For example, a conventional single-site alanine-scanning was used to assess the contribution of individual amino acid residues of a Fc fragment binding peptide displayed on filamentous phage [54]. Since this type of approach is laborious, methods have been developed for multiple alanine substitutions in a high-throughput manner [55]. One such approach builds on the codon-based mutagenesis, analyzing multiple positions, applying split and mix synthesis to produce degenerate oligonucleotides (one pool for the alanine codon and another for the wild-type codon) [56]. An alternative to alanine-scanning is serine-scanning, which follows the logic that, sometimes, substitutions with the hydrophobic alanine side chains may be more detrimental to the peptide’s affinity compared to the slightly larger but hydrophilic serine side chain. Similarly, homolog-scanning (substitutions at individual positions with similar residues) may be employed with the goal of minimizing structural disruption and identifying residues essential for maintaining a function [57].
Another site-directed mutagenesis type is the cassette mutagenesis. It works by replacing a section of genetic information with an alternative, synthetic sequence – a ‘cassette’ [58]. Different from other approaches that target short regions of a gene, this method is convenient for sequences up to 100 bp in size [59,60]. A prerequisite for this method to be practical is that the gene cassette must be flanked by two restriction sites that are complementary and unique with digest sites on the targeted vector. Restriction enzymes excise the targeted fragment from a vector that can then be replaced with DNA sequences carrying desired mutations. If a larger fragment is to be cloned, the ‘megaprimer’ approach is applied by amplification with a series of oligonucleotides [61]. This method can also benefit from using ‘spiked’ synthetic oligonucleotides, allowing randomization at multiple sites [62,63]. Cassette mutagenesis is based on Kunkel mutagenesis, which is time-consuming, so researchers developed an improved version termed ‘’PFunkel’’, a conflation of Pfu DNA polymerase and Kunkel mutagenesis, that can be performed in a day’s work [64,65]. To overcome the main constraint of site-directed mutagenesis, which is the tedious primer design, rational design techniques can be utilized to introduce desired mutations at precise positions. Researchers can leverage readily available tools such as AAscan, PCRdesign, and MutantChecker to simplify and boost the mutagenesis process [66].
Chemical-Based Mutagenesis
Chemical-based mutations involve various chemical methods to produce desired mutants. To chemically synthesize fully randomized oligonucleotides, a mixture of nucleotides must be applied at each coupling step [67]. A calamitous problem with this strategy is the pronounced bias resulting from the uneven incorporation frequency of the 4 nucleotide building blocks due to their inherent reactivity differences, rendering statistical random mutations inaccessible. Avoiding incorporation of stop codons is practically unattainable and the system is inclined towards amino acid residues encoded by redundant codons [68]. This problem can be tackled by adjusting the mutational frequency with ‘spiked oligonucleotides’ [62], taking into account the differences in reactivity of mononucleotides and the redundant genetic code. The essence of DNA spiking is that non-equimolar ratio of bases at targeted positions are applied during oligonucleotide synthesis, meaning each wild type nucleotide can be custom ‘doped’, achieving either ‘soft’ (high incidence of a certain nucleotide) or ‘hard’ (equal incidence of all four nucleotides) randomization, manually tuning the occurrence of certain amino acids at defined positions in the (poly)peptide chain.
Site-saturation mutagenesis seeks to achieve mutation at a maximal capacity by examining substitutions of a given residue against all possible amino acids. A fully randomized codon NNN (where N = A/C/G/T) gives rise to all possible 64 variant combinations (also known as 64-fold degeneracy) and codes for all 20 amino acids and 3 stop codons. This causes difficulties during library screening and risks enrichment of non-functional clones due to the random introduction of termination codons [69]. Operating with NNK, NNS, and NNB codons (where K = G/T, S = C/G, and B = C/G/T) minimizes the degeneracy in the third position of each codon, consequently lowering codon redundancy and the frequency of terminations [70]. However, such degenerate primers are expensive to synthesize, and using a single degenerate primer to completely eliminate codon redundancy while providing all 20 amino acids is unattainable, due to disproportional representation of certain amino acids [71,72]. Other strategies have to be employed to circumvent these constraints.
To synthesize redundancy-free mutagenic primers, mono [73], di [74], or trinucleotide phosphoramidite [75] solutions (or combinations [76]) can be used. This way, mixtures of oligonucleotides encoding all possible amino acid substitutions within a defined stretch of peptide or a limited number of amino acids (i.e., ‘tailored’ randomization) can be synthesized. This fine-tuning gives complete control over amino acid prevalence at defined positions in the corresponding (poly)peptide sequence, achieving ‘soft’ or ‘hard’ randomization. With this approach codon redundancy and stop codons are completely eliminated [68]. Another randomization strategy labeled ‘MAX’ eliminates genetic redundancy by using a collection of 20 primers containing only codons for each amino acid with the highest expression frequency in E. coli [77]. These primers are annealed to a template strand with completely randomized codons (NNN or NNK) at the targeted position. Any misannealing is trivial, since only the ligated selection strand is amplified by a subsequent PCR. The produced random cassettes are then enzyme-digested for cloning. Further development of this strategy gave birth to an upgraded version dubbed ProxiMAX in which multiple contiguous codons are randomized in a non-degenerate manner [78]. Here, a donor blunt-end dsDNA with terminal MAX codons and an upstream MlyI restriction site is ligated to an acceptor blunt-end dsDNA. The product strands are amplified, analyzed, and combined at desired ratios in the next randomization cycle. After each ligation cycle, endonuclease MlyI is applied to remove the donor DNA strand, making only the randomized sequences available for the successive ligation cycle.
Another strategy that has been developed by Tang et al. [71] is cost-effective and uses degenerate codons to eliminate or achieve near-zero redundancy. A mixture of four codons, NDT, VMA, ATG, and TGG (where D = A/G/T, V = A/C/G, M = A/C) with a molar ratio of 12:6:1:1 at each randomized position results in an equal theoretical distribution for each of the 20 amino acids, without occurrences of stop codons. Following a similar rationale, Kille et al. [72] developed the ‘’22c-trick’’ which uses only three codons per randomized position; NDT, VHG, and TGG (where H = A/C/T), at 12:9:1 molar ratio. The name sprung from the usage of 22 unique codons, achieving near uniform amino acid distribution (i.e., 2/22 for Leu and Val, and 1/22 for each of the remaining 18 amino acids). Other sophisticated primer mixing strategies have been reported [79–81], although picking the best approach is mostly dependent on the size and quality of the library to be prepared, and the lab’s operating budget [82].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21010215