Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Reproductive Biology
Infertility affects approximately 15% of couples worldwide of childbearing age, and in many cases the etiology of male infertility is unknown. The current standard evaluation of semen is insufficient to establish an accurate diagnosis. Spermatozoa cellular development and maturation are tightly coordinated by sperm protein phosphorylation, among other protein post-translational modifications (PTM). Proteomics techniques, such as phosphoproteomics, applied in this field are a powerful tool to understand the mechanisms that regulate sperm functions such as motility, which is essential for successful fertilization.
- human spermatozoa
- sperm proteins
- PTM
- phosphorylation
- phosphoproteomics
- male infertility
1. Introduction
Our understanding of sperm physiology remains relatively superficial despite the large number of manuscripts focused on spermatozoa [1]. Indeed, a global estimation calculates that 1 in 15 men of reproductive age are infertile, and the diagnosis of idiopathic male infertility, for which the cause is unknown, is a reality for 24% of men referred to assisted reproductive technology (ART) [2]. To date, basic semen analysis, or seminogram, is the best predictive test used routinely in laboratories for the assessment of male partner fertility, where it is analyzed if the semen samples meet the macroscopic (volume, pH, color, and viscosity) and microscopic characteristics (sperm concentration, total motility, progressive motility, and sperm morphology) established by the World Health Organization (WHO), which published a set of guidelines for evaluating semen quality 40 years ago. The last update, in 2010, includes the current reference parameters to make the first prognosis of male infertility [3].
Nevertheless, the seminogram is not the most suitable analysis to obtain an accurate diagnosis because semen parameters within the reference interval do not guarantee fertility, nor do values outside those limits necessarily imply male infertility or pathology [4]. Several studies have shown that men with sperm parameters (sperm number, morphology, and motility) below the thresholds outlined by the WHO can be fertile [5,6,7,8]. Additionally, in the same way, there are cases of men with normal sperm parameters that are infertile [9]. Therefore, more in-depth analysis and understanding of spermatozoa physiology at the molecular level are necessary to improve the current evaluation of male fertility by the routine semen analysis.
Fertilization might be considered the endpoint of sperm function. To get it successfully, spermatozoon, a highly specialized haploid cell that contains exceptionally condensed chromatin and will deliver the paternal DNA to the oocyte, must be completely functional. For that, spermatozoa undergo a series of physiological and biochemical changes from their developmental stages and during their transit through the male first and female reproductive tract later, which occur apparently in the complete absence of simultaneous gene transcription and protein translation. Although several coding and non-coding RNAs exist in human spermatozoa [10], which may play a role in gene silencing or heterochromatinization, their transcriptional and translational activities are nearly silent. Thus, sperm proteins of mature spermatozoa might undergo different post-translational modifications (PTM), such as phosphorylation or acetylation among, and become very important molecular mechanisms by which spermatozoa acquire functionality [11]. For this reason, the alteration of sperm status (for example, by errors in spermatogenesis or maturation) may be accompanied by a distinctive pattern of PTMs, characteristic of spermatozoa, in particular with low quality or motility and, therefore, low male reproductive prognostic.
In this regard, focusing our attention on sperm motile capacity, the abnormal content or presence of PTMs, including glutarylation, S-sulfhydration, hydroxyisobutyrylation and sumoylation of specific sperm proteins is associated with poor sperm motility, which indicates their relevant role in maintaining sperm motility [12,13,14,15]. Furthermore, asthenozoospermic men have shown a reduced abundance of lysine glutarylation in several proteins located in the tail of human spermatozoa, just as a diminished quantity of S-sulfhydrated H3 and H3.3 histones. These results positively correlate with sperm progressive motility [13,15]. On the other hand, the level of SUMO1-positive spermatozoa and the quantity of lysine 2-hydroxyisobutyrylation in sperm proteins trend towards higher grades in asthenozoospermic men compared with normozoospermic ones, indicating a negative association with the motility of human spermatozoa in this case [12,14]. Additionally, lysine acetylation seems to be essential for human sperm motility and fertilization [16].
The most extensively studied PTM in human spermatozoa is phosphorylation [17]. The phosphorylation of specific sperm proteins plays an important role in regulating sperm processes essential for fertilization, such as sperm motility, capacitation, or acrosome reactions [18,19,20]. Extraordinary advances have been achieved in the field of male infertility in recent decades, especially with the use of proteomics techniques and the bioinformatic analysis of human sperm proteomic data. However, there are many well-recognized causes of male infertility in humans whose molecular basis is only just beginning to be understood. The study of the global protein phosphorylation landscape of spermatozoa in different species proposes wide phosphoregulation in other processes such as sperm formation [21,22], maturation [23], capacitation [24,25,26,27,28], and motility [18,29,30,31,32]. Nevertheless, it remains to be fully explained which are ultimately the molecular mechanisms responsible for spermatozoa motility and, therefore, for sperm quality. Advances in global and quantitative methods to elucidate dynamic phosphorylation events in spermatozoa will be essential for a systematic understanding of their functional behavior. They will allow for a more comprehensive analysis of the biochemical basis of defective semen quality and identify possible biomarkers for different pathologies and conditions related to infertility. A few studies applying quantitative mass spectrometry (MS)-based proteomics have proposed some molecular mechanisms through which protein phosphorylation might affect sperm motility in humans [18,29,30,31].
2. Proteomics and Sperm Physiology
According to the recent data, the proteomic approach is a powerful tool to identify human sperm proteins as biomarkers of fertility [33]. For example, a recent seminal plasma proteomic-based study proposes the HSPA2 protein, a molecular chaperone mediating protein folding, as a possible biomarker of spermatogenesis status. Azoospermic men (who have a complete absence of spermatozoa in their ejaculate) lack HSPA2, which is present as additional protein isoforms in cryptozoospermia (<0.1 million spermatozoa mL−1) [34]. In addition, a study comparing high- and low-quality sperm nuclear extracts by proteomic analysis recently showed that the presence of Topoisomerase 2A in the human spermatozoa head is highly correlated to poor head morphology. So, Topoisomerase 2A, a protein normally involved in the alteration of DNA topology, may also be considered a potential biomarker to confirm male infertility in clinical practice [35]. The first study that examined the potential variability of the proteome in different semen samples and proposed proteomics as a useful tool for studying defects in sperm function was published almost 2 decades ago by Pixton and collaborators [36]. Since then, many proteomics studies have performed a comparative proteomics analysis between sperm cells from infertile patients and healthy donors [37,38,39,40,41,42,43,44,45,46].
On the other hand, sperm motility is essential for successful fertilization, so low sperm motility is highly associated with male infertility [47,48]. In fact, this defect has been the subject of research for years because it is frequently observed in andrology laboratories. In this sense, a retrospective study based on a large population reveals that about 82% of infertile men had impaired sperm motility [48]. Asthenozoospermia (AS), characterized by normal concentrations of spermatozoa (>15 million spermatozoa mL−1) and sperm progressive motility <32% [3], is one of the major causes of male infertility, which approximately accounts for 20% of infertility among men. The etiology of AS is varied and can be seen as a unique condition in isolated disease, associated with other sperm anomalies or as part of a syndromic association. In some cases, routine clinical examinations do not find clear causes, leading to so-called idiopathic asthenozoospermia [41]. Although the lower expression of several proteins might cause spermatozoa with poor motility, the molecular basis of AS is difficult to establish. Nonetheless, proteomic studies on asthenozoospermic individuals have increased in recent years, promoting the idea that the number of identified proteins related to sperm motility is rising [43].
For instance, in four different proteome analyses comparing sperm samples from asthenozoospermic vs. normozoospermic men, the altered expression of the HSPA2 protein was found. Interestingly, increased expression was observed in two studies [38,42], whereas HSPA2 expression decreased in the other two works [39,43]. Other chaperones HSPs (HSPA5, HSPA9, and HSPA1L) were also found downregulated in asthenozoospermic men [41,43]. Additionally, in a recent study comparing proteomes of high or low-motility human spermatozoa, HSPA1L and HSPA9 were also significantly decreased in low-motility spermatozoa [49]. Conversely, in other comparative sperm proteomics studies, the expression level of chaperones did not indicate significant differences [37,44,45,46]. It has to be mentioned that the lack of agreement or the opposite expression differences between the studies published may be due to several factors such as different quantitative technologies, different sample sources, low sample size, or even ethnic differences, among others. HSPs have a potential relationship with sperm quality, and they are important in normal sperm physiology [50], spermatogenesis, and sperm maturation [51,52,53], although the association between their altered expression and impaired motility is not yet fully understood. It is established that the role of the HSPs is to ensure the correct folding of proteins, their refolding of misfolded protein, and the orientation control of tagged proteins for subsequent degradation [54]. Thus, reduced HSPs expression might be associated with a decrease in sperm motility due to the accumulation of misfolded protein [55].
In addition, a higher level of triosephosphate isomerase (TPI), an extremely efficient metabolic enzyme in glycolysis and gluconeogenesis, is also associated with a reduced sperm motility phenotype [37,39,42]. Other proteins responsible for energy metabolism that could play an important role in spermatozoa motility maintenance are COX proteins (COX5B, COX6B, COX20, and COX41), which are involved in the oxidative phosphorylation (OXPHOS) pathway and showed lower levels in AS [38,42,43,45,46]. In conclusion, these are some examples that highlight the importance of glycolysis and OXPHOS as major metabolic pathways that provide energy to support human sperm motility.
Moreover, a reduction in sperm motility may be affected by other proteins, such as SEMG1 and SEMG2, which work as seminal plasma motility inhibitor proteins and are found up-regulated in asthenozoospermic men [37,38,46]. Another group of proteins with altered expression in spermatozoa with impaired motility involves different subunits of the proteasome such as PSMA3, PSMB3, PSMB4, PSMB5, PSMB6, PSMC2, PSMC6, and PSMD11 [37,38,39,42,45,49]. The proteasome plays a key role in the formation of condensed spermatozoa because it mediates the protein turnover of ubiquitinated proteins during spermatogenesis, when many proteins and organelles are degraded [55]. So, defects in the proteasome system might lead to the accumulation of ubiquitinated molecules and be related to sperm motility [39]. It is worth noting that SEMG1 and PSMB5 are also downregulated in a proteomic study that compares the proteomic profiles of human sperm samples that had or had not achieved a previous pregnancy via ART [56]. In addition, low levels of the major cytoskeleton components in spermatozoa flagella such as tektins (TEK1, TEK4, and TEK5) [39,41,42], outer dense fibers (ODF2) [41,43,49], or tubulin proteins (TUBB2C, TUBB2B, and TUBA3C) [39,43,49] are also associated with reduced sperm motility in comparative proteomics studies. On the other hand, altered levels of another protein that plays a role in the movement and structural organization of cells, such as CLU, have been found. CLU expression is decreased in some analyses [40,49], whereas it is increased in others [38,43,46].
Proteomics is also a current methodology used to study variations in sperm proteins that are altered in some disorders. For example, the SPEF2 protein is widely expressed in cilia-related organs such as the lung, spleen, trachea, brain, and testis [57], and its encoding gene is involved in a genetically heterogeneous disorder such as the so-called multiple morphological abnormalities of the sperm flagella (MMAF) [58]. Moreover, spermiogenesis failure by a deficiency in SPEF2 causes severe asthenoteratozoospermia, characterized by reduced sperm motility and abnormal sperm morphology [58]. Recently, an MS-proteomic analysis of human spermatozoa from three individuals with SPEF2 mutations compared with normal controls showed that this protein regulates the expression of various proteins involved in the flagellar assembly with which it interacts [59]. This methodology allows one to understand the protein networks from the whole sperm proteome, being especially useful in the study of sperm tail development since sperm flagellum is composed of more than 1000 proteins in the case of humans [60].
Human sperm cryopreservation plays an important role in assisted reproductive technology for male fertility preservation and the treatment of infertile couples. In this regard, proteomics approaches have also been useful to study the pathogenesis of sperm cryo-damage during the process of cryopreservation, comparing the proteomic differences between fresh and cryopreserved human sperm [61,62]. Fu’s lab, using MS and a novel proteomics technology named data-independent acquisition (DIA), identified 174 proteins significantly deregulated, including four enzymes involved in glycolysis (GPI, LDHB, ADH5, and PGAM1) and other proteins related to propanoate, glyoxylate, pyruvate, and dicarboxylate metabolism and gluconeogenesis [62]. Five years before, another proteomic analysis had found that 37% of the proteins involved in the metabolism are differentially expressed between freeze-thawed and fresh sperm samples [61]. So, both studies, although using different proteomic strategies, conclude that metabolic pathways play an important role during sperm cryo-preservation. Interestingly, phosphoglycerate mutase proteins, PGAM1 and PGAM2 [46] and the glucose 6-phosphate isomerase (GPI), evaluated by proteomic approaches, are also found to be significantly decreased in AS [42,46]. Furthermore, the supplement with the product of GPI, fructose-6-phosphate, significantly promotes human spermatozoa motility in vitro [46]. So, it can be postulated that during spermatozoa cryopreservation, when a marked reduction in sperm motility occurs, the supplement with fructose-6-phosphate could also help to recuperate the rates of spermatozoa motility.
Altogether, there are a set of proteins related to sperm quality in the literature. However, the internal relationship and the mechanisms underlying abnormal protein expressions and defective sperm function are not clear yet [49].
3. Phosphoproteomics Technique in Male Fertility
3.1. Phosphorylation as Post-Translational Modification of Sperm Proteins
Proteins can be edited after translation by PTMs in order to spread their functions and coordinate their signaling networks. Defects in PTMs have been linked to numerous human diseases and disorders, so the importance of PTMs in maintaining normal cellular states is essential [63]. Hence, previous and emerging data indicate that some male reproduction diseases, including the failure of sperm motility, arise through the deregulation of PTMs in spermatozoa. Despite that, more than 431 reversible and irreversible PTM mechanisms exist in the cell [64], including acetylation, methylation, glycosylation or ubiquitylation, among others, being protein phosphorylation the most widely studied post-translational modification of sperm proteins by far. We know that protein phosphorylation affects an estimated one-third of all cellular proteins [65], with most proteins phosphorylated at one or more sites in a mammalian cell [66]. However, we know only a small subset of the in vivo phosphorylation sites described. Most studies have focused on Ser, Thr, and Tyr phosphorylation (canonical phosphorylation), but there are other amino acid residues that are less common, including His, Lys, Arg, Asp, Glu, and Cys, that can also be phosphorylated (noncanonical phosphorylation) [67].This variability of possibilities further complicates the effort in studying protein phosphorylation. The consequence of covalent conjugation of phosphate groups to these residues frequently alters protein function by inducing conformational changes in proteins or by affecting protein-protein/enzyme-substrate interactions leading either to their activation or inactivation [68].
Nonetheless, protein phosphorylation is not permanent. The addition of a phosphoryl group to a target protein is a reversible modification determined by the activity of protein kinases and phosphatases on their substrates, which mediate protein phosphorylation and dephosphorylation processes, respectively. Both procedures give rise to conformational changes in proteins, have different triggers and remain consistently separated [69]. Consequently, the deregulation of kinases and phosphatases pathways is linked to many diseases, including infertility. So, deciphering the molecular elements that determine this biochemical balance is essential for correct reproductive function.
3.2. Phosphoproteomics Technique
Phosphoproteomics is a large-scale analysis that identifies and quantifies the phosphorylated proteins in addition to the mapping of the phosphorylation sites in a complex biological sample using MS [70]. Briefly, an MS-based phosphoproteomics study on the role of in vivo phosphorylation in sperm physiology starts with isolating sperm cells from the seminal plasma and other cells coexisting in semen by swim-up procedure, density centrifugation, or different techniques. The purity of the sperm preparation and the removal of interfering compounds are critical steps in the process because any minor contamination could result in a false-positive identification [71]. Sperm proteins are then extracted and protein mixtures are digested with a specific protease, typically trypsin.
Once proteins are extracted, carrying out a phosphopeptides enrichment procedure before experimental analysis is necessary, given that, for example, almost 30% of all human proteins may be phosphorylated and that each phosphoprotein may exist as multiple phospho-isoforms with different relative abundances and stoichiometries [70]. In this sense, a wide variety of studies searched for increased sensitivity, indispensable to avoid the suppression of phosphorylated peptides caused by the effect on the ionization of the phosphate group in MS, which result in decreased signal intensity for phosphorylated peptides in the presence of non-phosphorylated ones [72]. Among the wide selection of methodologies developed for phosphopeptides enrichment, the most extensively used in the study of sperm cells is immobilized metal ion affinity chromatography (IMAC) [17], which is based upon the affinity that phosphate exhibits towards immobilized metal ions and forms relatively stable complexes with these. So, the nature of the chromatographic stationary phase is of extreme importance [73]. Accordingly, titanium dioxide (TiO2) resin has been one of the most widespread methods for phosphopeptides enrichment from complex biological samples because it has a very high affinity for phosphopeptides, is extremely tolerant towards most buffers used in biological experiments, and is optimal for large-scale phosphoproteomics studies [74]. Furthermore, TiO2 microcolumns significantly enhance the binding selectivity of TiO2 toward phosphorylated peptides, thereby enabling phosphorylated peptide characterization from low femtomole level phosphorylated proteins and improving selectivity by reducing unspecific binding of non-phosphorylated peptides [72].
Later, the sperm phosphopeptides are detected using both conventional and advanced proteomic techniques. Two-dimensional (2D) gel electrophoresis separates sperm proteins based on peptides’ isoelectric focusing properties and molecular weight. A modified version named difference gel electrophoresis (DIGE) identifies differentially expressed proteins (DEPs) [75]. The analysis of advanced high-throughput techniques such as MALDI-TOF (matrix-assisted laser desorption/ionization time-of-flight) and LC-MS/MS (liquid chromatography-tandem mass spectrometry) detect low abundance peptides present in a sample with low protein concentration. Therefore, they overcome the limitations of conventional proteomics techniques. In addition, advances in chromatography techniques such as nano HPLC (high-performance liquid chromatography) or UPLC (ultra-performance liquid chromatography) methods enable the decrease of the internal diameter of the LC column to analyze low amounts of a sample with none or very low dilution and with increased sensitivity, which allows for higher sample throughput [73]. Those aspects are fundamental in phosphoproteomics studies.
In addition, MS offers numerous advantages for studying protein phosphorylation, enabling its quantitative, sensitive, and site-specific measure [76]. MS-based quantification strategies rely on light/heavy peptide intensities and can be divided into label-based and label-free approaches. Label-based quantitation methods utilize stable isotope labels by chemical, metabolic, or proteolytic labeling strategies. These are incorporated within the peptides, introducing an expectable mass difference within the two or more experimental conditions. The quantitation is based on comparing the peak intensity ratio of the labeled peptide pairs [77]. In sperm phosphoproteome studies, tandem mass tag (TMT) is the most widely used peptides labeling technique [28,30,32]. In this strategy, the peptides are labeled with chemical groups which are isobaric (identical in mass) in nature but dissociate under tandem MS to yield reporter ions of variable mass [77]. In contrast, label-free quantitation compares both relative and absolute protein quantity by utilizing signal intensity and spectral counting of the same peptide [77]. It is also one of the methods of choice in human sperm phosphoproteome studies [27,31] and has gained more acceptance because it shows the highest proteome coverage and is cost-efficient without adding additional steps to labeling samples with alternative differential mass tags [78].
Finally, mass spectral data interpretation is carried out using the different platforms, databases, and software programs available, which allow for the identification and quantification of the assignments of peptides and proteins that make up the sperm phosphoproteome. Bioinformatics methods are indispensable for proteomics-based studies and are helping scientists to interpret the integration of large datasets from proteomics studies [79]. The specific workflow involving the processing of semen samples for sperm phosphoproteomics analysis is shown in Figure 1.
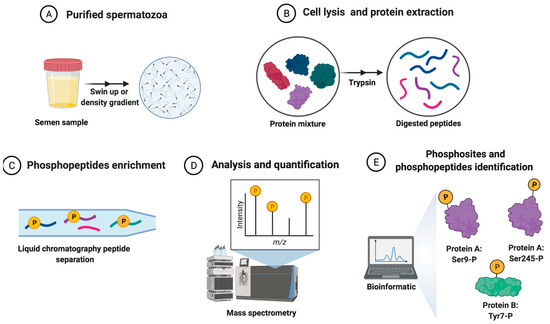
Figure 1. The general workflow of the quantitative phosphoproteomics strategy for human spermatozoa samples analysis from sperm donors. (A) Purified sperm cells from semen samples (spermatozoa isolated from other cells and the seminal plasma). (B) Sperm protein extraction and digestion after cellular lysis. (C) Phosphopeptides enrichment and peptide separation. (D) The analysis and quantification of peptides. (E) The collection and analysis of sperm phosphoproteome data by bioinformatics tools.
This entry is adapted from the peer-reviewed paper 10.3390/biology11050659
This entry is offline, you can click here to edit this entry!