Despite advances in its treatment, heart failure remains a major cause of morbidity and mortality, evidencing an urgent need for novel mechanism-based targets and strategies. Myocardial hypertrophy, caused by a wide variety of chronic stress stimuli, represents an independent risk factor for the development of heart failure, and its prevention constitutes a clinical objective. Recent studies performed in preclinical animal models support the contribution of the Ca2+-dependent cysteine proteases calpains in regulating the hypertrophic process and highlight the feasibility of their long-term inhibition as a pharmacological strategy.
- calpain
- NFAT Pathway
- Ca2+
1. Pro-Hypertrophic Pathways Modulated by Calpains
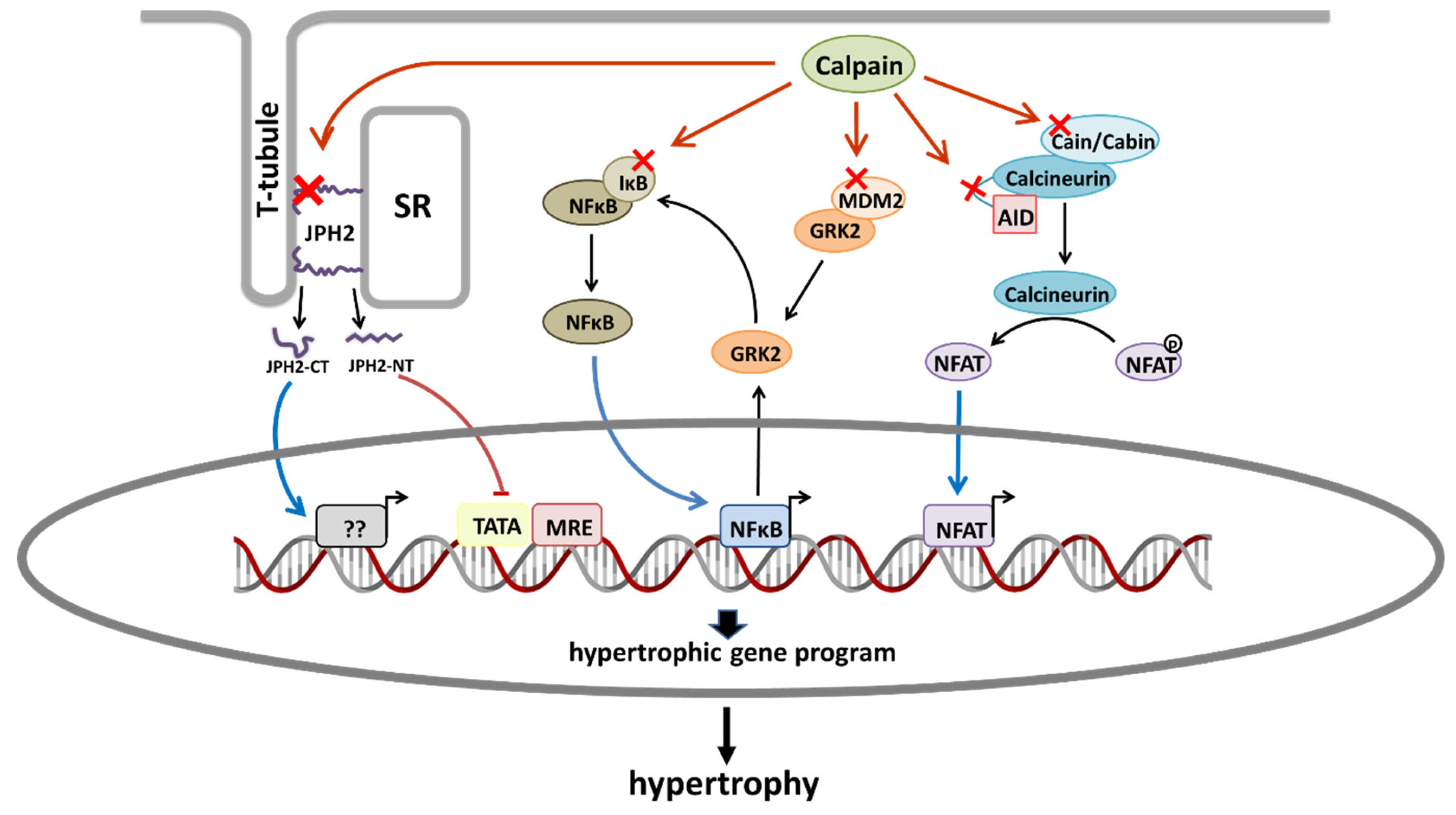
1.1. Calmodulin/NFAT Pathway
1.2. NF-κB Activation
1.3. GRK2 Upregulation
1.4. Junctophilin-2 Cleavage
2. Calpain Contribution to the Progression of Pathologic Hypertrophy
2.1. Proteolysis of Myosin Light Chain Kinase
2.2. Mitochondrial Damage
3. Pharmacological Inhibition of Calpains
This entry is adapted from the peer-reviewed paper 10.3390/ijms23084103
References
- Winkle, A.J.; Nassal, D.M.; Shaheen, R.; Thomas, E.; Mohta, S.; Gratz, D.; Weinberg, S.H.; Hund, T.J. Emerging therapeutic targets for cardiac hypertrophy. Expert Opin. Ther. Targets 2022, 26, 29–40.
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407.
- Chaklader, M.; Rothermel, B.A. Calcineurin in the heart: New horizons for an old friend. Cell. Signal. 2021, 87, 110134.
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT Coupling Participates in Pathological, but not Physiological, Cardiac Hypertrophy. Circ. Res. 2004, 94, 110–118.
- Burkard, N.; Becher, J.; Heindl, C.; Neyses, L.; Schuh, K.; Ritter, O. Targeted proteolysis sustains calcineurin activation. Circulation 2005, 111, 1045–1053.
- Kim, M.J.; Jo, D.G.; Hong, G.S.; Kim, B.J.; Lai, M.; Cho, D.H.; Kim, K.W.; Bandyopadhyay, A.; Hong, Y.M.; Kim, D.H.; et al. Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc. Natl. Acad. Sci. USA 2002, 99, 9870–9875.
- Li, Y.; Ma, J.; Zhu, H.; Singh, M.; Hill, D.; Greer, P.A.; Arnold, J.M.; Abel, E.D.; Peng, T. Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes 2011, 60, 2985–2994.
- Letavernier, E.; Perez, J.; Bellocq, A.; Mesnard, L.; De Castro Keller, A.; Haymann, J.P.; Baud, L. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ. Res. 2008, 102, 720–728.
- Kawano, S.; Kubota, T.; Monden, Y.; Tsutsumi, T.; Inoue, T.; Kawamura, N.; Tsutsui, H.; Sunagawa, K. Blockade of NF-κB improves cardiac function and survival after myocardial infarction. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H1337–H1345.
- Kawano, S.; Kubota, T.; Monden, Y.; Kawamura, N.; Tsutsui, H.; Takeshita, A.; Sunagawa, K. Blockade of NF-κB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc. Res. 2005, 67, 689–698.
- Muller, D.N.; Dechend, R.; Mervaala, E.M.A.; Park, J.K.; Schmidt, F.; Fiebeler, A.; Theuer, J.; Breu, V.; Ganten, D.; Haller, H.; et al. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 2000, 35, 193–201.
- Hong, H.Q.; Lu, J.; Fang, X.L.; Zhang, Y.H.; Cai, Y.; Yuan, J.; Liu, P.Q.; Ye, J.T. G3BP2 is involved in isoproterenol-induced cardiac hypertrophy through activating the NF-κB signaling pathway. Acta Pharmacol. Sin. 2018, 39, 184–194.
- Ye, T.; Wang, Q.; Zhang, Y.; Song, X.; Yang, D.; Li, D.; Li, D.; Su, L.; Yang, Y.; Ma, S. Over-expression of calpastatin inhibits calpain activation and attenuates post-infarction myocardial remodeling. PLoS ONE 2015, 10, e0120178.
- Aluja, D.; Inserte, J.; Penela, P.; Ramos, P.; Ribas, C.; Iñiguez, M.Á.; Mayor, F.; Garcia-Dorado, D. Calpains mediate isoproterenol-induced hypertrophy through modulation of GRK2. Basic Res. Cardiol. 2019, 114, 21.
- Poncelas, M.; Inserte, J.; Aluja, D.; Hernando, V.; Vilardosa, U.; Garcia-Dorado, D. Delayed, oral pharmacological inhibition of calpains attenuates adverse post-infarction remodelling. Cardiovasc. Res. 2017, 113, 950–961.
- Freund, C.; Schmidt-Ullrich, R.; Baurand, A.; Dunger, S.; Schneider, W.; Loser, P.; El-Jamali, A.; Dietz, R.; Scheidereit, C.; Bergmann, M.W. Requirement of nuclear factor-κB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation 2005, 111, 2319–2325.
- Shumway, S.D.; Maki, M.; Miyamoto, S. The PEST domain of IκBα is necessary and sufficient for in vitro degradation by μ-calpain. J. Biol. Chem. 1999, 274, 30874–39881.
- Han, Q.; Liu, Q.; Zhang, H.; Lu, M.; Wang, H.; Tang, F.; Zhang, Y. Simvastatin improves cardiac hypertrophy in diabetic rats by attenuation of oxidative stress and inflammation induced by calpain1-mediated activation of nuclear factor-κb (Nf-κb). Med. Sci. Monit. 2019, 25, 1232–1241.
- Arcones, A.C.; Murga, C.; Penela, P.; Inserte, J.; Mayor, F. G protein–coupled receptor kinase 2 at crossroads of metabolic and cardiovascular diseases. Curr. Opin. Endocr. Metab. Res. 2021, 16, 75–85.
- Penela, P.; Ribas, C.; Sánchez-Madrid, F.; Mayor, F. G protein-coupled receptor kinase 2 (GRK2) as a multifunctional signaling hub. Cell. Mol. Life Sci. 2019, 76, 4423–4446.
- Lieu, M.; Koch, W.J. GRK2 and GRK5 as therapeutic targets and their role in maladaptive and pathological cardiac hypertrophy. Expert Opin. Ther. Targets 2019, 23, 201–214.
- Schlegel, P.; Reinkober, J.; Meinhardt, E.; Tscheschner, H.; Gao, E.; Schumacher, S.M.; Yuan, A.; Backs, J.; Most, P.; Wieland, T.; et al. G protein-coupled receptor kinase 2 promotes cardiac hypertrophy. PLoS ONE 2017, 12, e0182110.
- Sorriento, D.; Santulli, G.; Franco, A.; Cipolletta, E.; Napolitano, L.; Gambardella, J.; Gomez-Monterrey, I.; Campiglia, P.; Trimarco, B.; Iaccarino, G.; et al. Integrating GRK2 and NFkappaB in the Pathophysiology of Cardiac Hypertrophy. J. Cardiovasc. Transl. Res. 2015, 8, 493–502.
- Penela, P. Chapter Three—Ubiquitination and Protein Turnover of G-Protein-Coupled Receptor Kinases in GPCR Signaling and Cellular Regulation; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 141, ISBN 9780128093863.
- Chen, B.; Guo, A.; Zhang, C.; Chen, R.; Zhu, Y.; Hong, J.; Kutschke, W.; Zimmerman, K.; Weiss, R.M.; Zingman, L.; et al. Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc. Res. 2013, 100, 54–62.
- Wang, Y.; Chen, B.; Huang, C.K.; Guo, A.; Wu, J.; Zhang, X.; Chen, R.; Chen, C.; Kutschke, W.; Weiss, R.M.; et al. Targeting Calpain for Heart Failure Therapy: Implications From Multiple Murine Models. JACC Basic Transl. Sci. 2018, 3, 503–517.
- Murphy, R.M.; Dutka, T.L.; Horvath, D.; Bell, J.R.; Delbridge, L.M.; Lamb, G.D. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J. Physiol. 2013, 591, 719–729.
- Guo, A.; Hall, D.; Zhang, C.; Peng, T.; Miller, J.D.; Kutschke, W.; Grueter, C.E.; Johnson, F.L.; Lin, R.Z.; Song, L.S. Molecular determinants of calpain-dependent cleavage of junctophilin-2 protein in cardiomyocytes. J. Biol. Chem. 2015, 290, 17946–17955.
- Guo, A.; Wang, Y.; Chen, B.; Wang, Y.; Yuan, J.; Zhang, L.; Hall, D.; Wu, J.; Shi, Y.; Zhu, Q.; et al. E-C coupling structural protein junctophilin-2 encodes a stress- adaptive transcription regulator. Science 2018, 362, eaan3303.
- Lahiri, S.K.; Quick, A.P.; Samson-Couterie, B.; Hulsurkar, M.; Elzenaar, I.; van Oort, R.J.; Wehrens, X.H.T. Nuclear localization of a novel calpain-2 mediated junctophilin-2 C-terminal cleavage peptide promotes cardiomyocyte remodeling. Basic Res. Cardiol. 2020, 115, 49.
- Wang, J.; Ciampa, G.; Zheng, D.; Shi, Q.; Chen, B.; Dale Abel, E.; Peng, T.; Hall, D.D.; Song, L.S. Calpain-2 specifically cleaves Junctophilin-2 at the same site as Calpain-1 but with less efficacy. Biochem. J. 2021, 478, 3539–3553.
- Huang, J.; Shelton, J.M.; Richardson, J.A.; Kamm, K.E.; Stull, J.T. Myosin regulatory light chain phosphorylation attenuates cardiac hypertrophy. J. Biol. Chem. 2008, 283, 19748–19756.
- Warren, S.A.; Briggs, L.E.; Zeng, H.; Chuang, J.; Chang, E.I.; Terada, R.; Li, M.; Swanson, M.S.; Lecker, S.H.; Willis, M.S.; et al. Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation 2012, 126, 2575–2588.
- Wang, S.; Wang, H.; Su, X.; Liu, B.; Wang, L.; Yan, H.; Mao, S.; Huang, H.; Huang, C.; Cheng, M.; et al. β-adrenergic activation may promote myosin light chain kinase degradation through calpain in pressure overload-induced cardiac hypertrophy: β-adrenergic activation results in MLCK degradation. Biomed. Pharmacother. 2020, 129, 110438.
- Arrington, D.D.; Van Vleet, T.R.; Schnellmann, R.G. Calpain 10: A mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am. J. Physiol.-Cell Physiol. 2006, 291, 1159–1171.
- Chen, Q.; Paillard, M.; Gomez, L.; Ross, T.; Hu, Y.; Xu, A.; Lesnefsky, E.J. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem. Biophys. Res. Commun. 2011, 415, 533–538.
- Shintani-Ishida, K.; Yoshida, K.I. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int. J. Cardiol. 2015, 197, 26–32.
- Zhang, M.; Wang, G.; Peng, T. Calpain-Mediated Mitochondrial Damage: An Emerging Mechanism Contributing to Cardiac Disease. Cells 2021, 10, 2024.
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106.
- Chen, M.; He, H.; Zhan, S.; Krajewski, S.; Reed, J.C.; Gottlieb, R.A. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem. 2001, 276, 30724–30728.
- Luo, T.; Yue, R.; Hu, H.; Zhou, Z.; Yiu, K.H.; Zhang, S.; Xu, L.; Li, K.; Yu, Z. PD150606 protects against ischemia/reperfusion injury by preventing μ-calpain-induced mitochondrial apoptosis. Arch. Biochem. Biophys. 2015, 586, 1–9.
- Khan, H.; Garg, N.; Singh, T.G.; Kaur, A.; Thapa, K. Calpain Inhibitors as Potential Therapeutic Modulators in Neurodegenerative Diseases. Neurochem. Res. 2022, 47, 1125–1149.
- Inserte, J.; Cardona, M.; Poncelas-Nozal, M.; Hernando, V.; Vilardosa, Ú.; Aluja, D.; Parra, V.M.; Sanchis, D.; Garcia-Dorado, D. Studies on the role of apoptosis after transient myocardial ischemia: Genetic deletion of the executioner caspases-3 and -7 does not limit infarct size and ventricular remodeling. Basic Res. Cardiol. 2016, 111, 18.
- Chen, Q.; Thompson, J.; Hu, Y.; Dean, J.; Lesnefsky, E.J. Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion. Am. J. Physiol. Cell Physiol. 2019, 317, C910–C921.
- Ni, R.; Zheng, D.; Xiong, S.; Hill, D.J.; Sun, T.; Gardiner, R.B.; Fan, G.C.; Lu, Y.; Abel, E.D.; Greer, P.A.; et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: A novel mechanism contributing to diabetic cardiomyopathy. Diabetes 2016, 65, 255–268.
- Cao, T.; Fan, S.; Zheng, D.; Wang, G.; Yu, Y.; Chen, R.; Song, L.S.; Fan, G.C.; Zhang, Z.; Peng, T. Increased calpain-1 in mitochondria induces dilated heart failure in mice: Role of mitochondrial superoxide anion. Basic Res. Cardiol. 2019, 114, 17.
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022.
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2021, 178, 2060–2076.
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.L.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J. Biol. Chem. 2015, 290, 168–182.
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116.
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268.
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542.
- Guan, L.; Che, Z.; Meng, X.; Yu, Y.; Li, M.; Yu, Z.; Shi, H.; Yang, D.; Yu, M. MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy Inhibition. J. Cell. Mol. Med. 2019, 23, 7830–7843.
- Ono, Y.; Saido, T.C.; Sorimachi, H. Calpain research for drug discovery: Challenges and potential. Nat. Rev. Drug Discov. 2016, 15, 854–876.
- Letavernier, E.; Zafrani, L.; Perez, J.; Letavernier, B.; Haymann, J.P.; Baud, L. The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res. 2012, 96, 38–45.
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal inflammation, cell death and inherited retinal dystrophies. Int. J. Mol. Sci. 2021, 22, 2096.
- Kumamoto, T.; Ueyama, H.; Sugihara, R.; Kominami, E.; Goll, D.E.; Tsuda, T. Calpain and cathepsins in the skeletal muscle of inflammatory myopathies. Eur. Neurol. 1997, 37, 176–181.
- Nian, H.; Ma, B. Calpain–calpastatin system and cancer progression. Biol. Rev. 2021, 96, 961–975.
- Dókus, L.E.; Yousef, M.; Bánóczi, Z. Modulators of calpain activity: Inhibitors and activators as potential drugs. Expert Opin. Drug Discov. 2020, 15, 471–486.
- Arthur, G.D.; Belcastro, A.N. A calcium stimulated cysteine protease involved in isoproterenol induced cardiac hypertrophy. Mol. Cell. Biochem. 1997, 176, 241–248.
- Mehdi, S. Cell-penetrating inhibitors of calpain. Trends Biochem. Sci. 1991, 16, 150–153.
- Khalil, P.N.; Neuhof, C.; Huss, R.; Pollhammer, M.; Khalil, M.N.; Neuhof, H.; Fritz, H.; Siebeck, M. Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/reperfusion model. Eur. J. Pharmacol. 2005, 528, 124–131.
- Jantos, K.; Kling, A.; Mack, H.; Hornberger, W.; Moeller, A.; Nimmrich, V.; Lao, Y.; Nijsen, M. Discovery of ABT-957: 1-Benzyl-5-oxopyrrolidine-2-carboxamides as selective calpain inhibitors with enhanced metabolic stability. Bioorganic Med. Chem. Lett. 2019, 29, 1968–1973.
- Lon, H.K.; Mendonca, N.; Goss, S.; Othman, A.A.; Locke, C.; Jin, Z.; Rendenbach-Mueller, B. Pharmacokinetics, Safety, Tolerability, and Pharmacodynamics of Alicapistat, a Selective Inhibitor of Human Calpains 1 and 2 for the Treatment of Alzheimer Disease: An Overview of Phase 1 Studies. Clin. Pharmacol. Drug Dev. 2018, 8, 290–303.
- Shirasaki, Y.; Yamaguchi, M.; Miyashita, H. Retinal penetration of calpain inhibitors in rats after oral administration. J. Ocul. Pharmacol. Ther. 2006, 22, 417–424.
- Yoshikawa, Y.; Zhang, G.X.; Obata, K.; Ohga, Y.; Matsuyoshi, H.; Taniguchi, S.; Takaki, M. Cardioprotective effects of a novel calpain inhibitor SNJ-1945 for reperfusion injury after cardioplegic cardiac arrest. Am. J. Physiol.-Heart Circ. Physiol. 2010, 298, H643–H651.
- Pollack, J.R.; Witt, R.C.; Sugimoto, J.T. Differential effects of calpain inhibitors on hypertrophy of cardiomyocytes. Mol. Cell. Biochem. 2003, 251, 47–50.
- Sheng, J.J.; Chang, H.; Yu, Z. Bin Nuclear Translocation of Calpain-2 Mediates Apoptosis of Hypertrophied Cardiomyocytes in Transverse Aortic Constriction Rat. J. Cell. Physiol. 2015, 230, 2743–2754.
- Wang, K.K.W.; Nath, R.; Posner, A.; Raser, K.J.; Buroker-Kilgore, M.; Hajimohammadreza, I.; Probert, J.; Marcoux, F.W.; Ye, Q.; Takano, E.; et al. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell- permeable calpain inhibitor and is neuroprotective. Proc. Natl. Acad. Sci. USA 1996, 93, 6687–6692.
- Shinkai-Ouchi, F.; Koyama, S.; Ono, Y.; Hata, S.; Ojima, K.; Shindo, M.; DuVerle, D.; Ueno, M.; Kitamura, F.; Doi, N.; et al. Predictions of cleavability of calpain proteolysis by quantitative structure-activity relationship analysis using newly determined cleavage sites and catalytic efficiencies of an oligopeptide array. Mol. Cell. Proteom. 2016, 15, 1262–1280.
- Santos, D.M.; Xavier, J.M.; Morgado, A.L.; Solá, S.; Rodrigues, C.M.P. Distinct regulatory functions of calpain 1 and 2 during neural stem cell self-renewal and differentiation. PLoS ONE 2012, 7, e33468.
- Wang, Y.; Lopez, D.; Davey, P.G.; Cameron, D.J.; Nguyen, K.; Tran, J.; Marquez, E.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol. Dis. 2016, 93, 121–128.
- Ozaki, T.; Yamashita, T.; Ishiguro, S.I. Ca2+-induced release of mitochondrial m-calpain from outer membrane with binding of calpain small subunit and Grp75. Arch. Biochem. Biophys. 2011, 507, 254–261.
- Ozaki, T.; Nakazawa, M.; Yamashita, T.; Sorimachi, H.; Hata, S.; Tomita, H.; Isago, H.; Baba, A.; Ishiguro, S.-I. Intravitreal injection or topical eye-drop application of a μ-calpain C2L domain peptide protects against photoreceptor cell death in Royal College of Surgeons’ rats, a model of retinitis pigmentosa. Biochim. Biophys. Acta 2012, 1822, 1783–1795.
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2013, 55, 31–41.
- Facundo, H.D.T.F.; Brainard, R.E.; Caldas, F.R.D.L.; Lucas, A.M.B. Mitochondria and cardiac hypertrophy. Adv. Exp. Med. Biol. 2017, 982, 203–226.
- Gong, X.; Yu, Z.; Huang, Z.; Xie, L.; Zhou, N.; Wang, J.; Liang, Y.; Qin, S.; Nie, Z.; Wei, L.; et al. Protective effects of cardiac resynchronization therapy in a canine model with experimental heart failure by improving mitochondrial function: A mitochondrial proteomics study. J. Interv. Card. Electrophysiol. 2021, 61, 123–135.