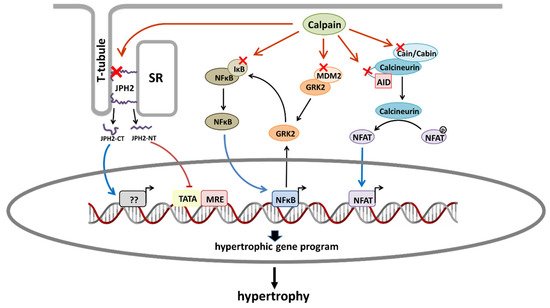
1. Pro-Hypertrophic Pathways Modulated by Calpains
Development of pathologic cardiac hypertrophy involves a vast network of receptors, signaling pathways, and effector proteins that result in the activation of transcription factors, which, in turn, activate pro-hypertrophic gene expression programs. Excellent reviews covering the general topic have been written [
112,
113]. Here, instead, we focus on those signaling pathways that have been proposed to be regulated by calpains (
Figure 1).
Figure 1. Schematic diagram showing the main proposed mechanisms by which calpains promote cardiac hypertrophy. Red crosses indicate calpain substrates that are involved in hypertrophic signaling pathways. From left to right: Calpain-2-dependent proteolysis of JPH2 generates a JPH2-CT fragment that translocates to the nucleus and favors hypertrophy. Calpain-1-dependent proteolysis of JPH2 produces a JPH2-NT fragment that acts as a stress-adaptive transcription regulator preventing hypertrophy. Calpain-dependent degradation of IkBα activates NFκB. Calpain activity promotes the upregulation of GRK2 by mechanisms affecting both its stability (degradation of MDM2) and transcription (activation of NFκB). GRK2 overexpression phosphorylates IκBα promoting its proteosomal degradation and the subsequent activation of NFκB. Proteolysis of cain/cabin or calcineurin AID induces the activation of NFAT. AID, autoinhibitory domain; JPH2, junctophilin 2; JPH2-CT, junctophilin 2 C-terminal fragment; JPH2-NT; SR, sarcoplasmic reticulum.
1.1. Calmodulin/NFAT Pathway
In addition to calpains, intracellular Ca
2+ dysregulation is secondary to chronic stress, which also results in the activation of other Ca
2+-dependent enzymes, such as calcineurin. Calcineurin is a Ca
2+ and calmodulin sensitive phosphatase which, among other functions, plays a master regulatory role in the hypertrophic response of cardiomyocytes to a chronic stimulus by activating the transcription factor NFAT [
114]. Further, activation of the calcineurin/NFAT pathway has been proposed to participate in pathological but not physiological forms of cardiac hypertrophy [
115]. Interestingly, different studies suggest that calpain activation can modulate this signaling cascade by acting at different levels. Angiotensin II stimulation of cardiomyocytes has been shown to produce calpain-dependent proteolysis of the autoinhibitory domain of calcineurin resulting in a constitutive nuclear form, which remains active even after removal of the hypertrophic stimulus [
116]. In addition, in vitro studies propose that calpain-1 may also activate calcineurin by cleaving the calcineurin-binding domain of the endogenous calcineurin inhibitor cain/cabin [
117]. Consistent with this scenario, the attenuation of calpain activity by constitutive calpastatin overexpression or Capn4 genetic deletion reduced the activation of the NFAT pathway and attenuated myocardial hypertrophy in a mouse model of type 1 diabetes [
101]. By contrast, inhibition of calpain by using the same calpastatin overexpressing mouse strain prevented cardiac hypertrophy induced by chronic infusion of angiotensin II through a mechanism independent of NFAT activation but dependent on the translocation to the nucleus of NF-κB [
39].
1.2. NF-κB Activation
A solid body of evidence supports a critical role of the nuclear factor NF-κB in cardiac hypertrophy induced by a wide variety of chronic pathologic stimuli. Targeted disruption of the p50 NF-κB subunit reduced cardiomyocyte hypertrophy and improved cardiac function after MI [
118], while deletion of its c-Rel subunit ameliorated cardiac hypertrophy in response to chronic infusion of angiotensin II [
118,
119]. In line with these studies, the administration of the NF-κB inhibitor, PDTC, reduced cardiac hypertrophy resulting from angiotensin II [
120] or isoproterenol treatments [
121].
Accumulating evidence obtained by different groups convincingly demonstrates that calpain overactivation enhances NF-κB activity. Calpain inhibition by genetic deletion of Capn4 or calpastatin overexpression prevents the nuclear translocation of the p65 NF-κB subunit and attenuates hypertrophy induced by myocardial ischemia and angiotensin II [
39,
89]. Similar results have been obtained by using calpain inhibitors in models of MI with transient LAD ligation or isoproterenol administration [
38,
40].
NF-κB is sequestered in the cytoplasm by the interaction with its inhibitory protein IκBα. The expression of a mutant IκBα that acts as a super-repressor of NF-κB in transgenic mice attenuates hypertrophy induced by isoproterenol or angiotensin II infusion [
122], pointing to IκBα degradation as a necessary step for the nuclear translocation of the p65 NF-κB subunit and the progress of pathological hypertrophy. Although canonical IκBα proteolysis involves the ubiquitin-proteasome pathway, IκBα is a well-known calpain substrate [
123] and its calpain-dependent cleavage promotes hypertrophy in response to MI [
40], angiotensin II [
122], adrenergic stimulation [
38], and in streptozotocin-induced diabetic rats [
124]. Altogether, these studies demonstrate that calpains contribute to cardiomyocyte hypertrophy at least in part by activating NF-κB through the direct proteolysis of IκBα.
1.3. GRK2 Upregulation
Angiotensin II, endothelin-1, phenylephrine, and isoproterenol are well documented, showing cardiac hypertrophy by activating pathways linked to G protein-coupled receptors (GPCRs). G protein-coupled receptor kinases (GRKs) are key modulators of GPCRs in physiological and pathological conditions and have attracted a lot of attention primarily for their role in regulating β-adrenergic receptors in the context of cardiac contraction [
125,
126]. In addition, different groups provide evidence of non-canonical roles of the GRK2 isoform, including its ability to modulate cardiac hypertrophy [
127]. GRK2 is upregulated in patients with HF and preclinical models of chronic stress of either hypertensive or ischemic origin [
38,
128]. In H9c2 cells, a rat myoblast cell line, the overexpression of GRK2 is enough to elicit a hypertrophic response [
129], while its genetic deletion attenuates hypertrophy induced by TAC or isoproterenol administration [
38,
128]. A recent study from our group demonstrates that calpain activation in response to chronic isoproterenol administration promotes the overexpression of GRK2 by mechanisms affecting both its stability and transcription [
38]. Isoproterenol treatment induces a calpain-dependent decrease of cardiac MDM2 levels, the major E3 ligase implicated in the ubiquitination and degradation of GRK2 [
130], thus enhancing GRK2 stability. Moreover, the GRK2 promoter sequence has several canonical binding sites for NF-κB. Isoproterenol-induced calpain activation cleavages IκBα, leading to NF-κB translocation, which enhances the transcriptional activity of the GRK2 promoter. Further, it has been proposed that GRK2 can also phosphorylate IκBα, favoring its proteasomal degradation and the subsequent activation of NF-κB [
129], suggesting that myocardial GRK2 and NF-κB co-regulate each other to trigger hypertrophic gene transcriptional activation. Remarkably, chronic administration of an oral calpain inhibitor prevented isoproterenol-dependent GRK2 upregulation, while hemizygous GRK2 mice showed attenuated myocardial hypertrophy. Overall, these studies strongly suggest that the calpain-dependent modulation of the MDM2/GRK2 axis is a relevant event in cardiac hypertrophy downstream calpain overactivation.
1.4. Junctophilin-2 Cleavage
Recently, the calpain-dependent proteolysis of junctophilin-2 (JPH2) has emerged as a novel mechanism involved in the regulation of cardiomyocyte growth. Junctophilin-2, a structural protein connecting T-tubules and the sarcoplasmic reticulum, is essential for maintaining normal T-tubule organization and an efficient excitation–contraction coupling in adult cardiomyocytes [
131]. Different studies demonstrate that calpain activation induced by several models of cardiac stress, including MI with permanent LAD occlusion, TAC, and isoproterenol infusion, results in the cleavage of JPH2 and the disruption of the contractile machinery, driving HF progression [
41,
132,
133]. The correlation between the reduction of JPH2 levels and increased calpain activity has also been confirmed in failing human hearts [
41]. In addition to its effects on contractility, it has been recently suggested that calpain-1 cleaves JPH2 at a conserved R565/T566 site and the resulting JPH2 N-terminal fragment (JPH2NT) translocates to the nucleus, where it acts as a stress-adaptive transcription regulator through Mef2 gene repression [
134]. Supporting this role of JPH2NT, the transgenic overexpression of JPH2NT attenuated pathological remodeling in response to TAC, while genetic mice with a loss of function of JP2NT exacerbated hypertrophy and cardiac dysfunction. More recently, the nuclear localization of a novel C-terminal fragment (JPH2CT) generated by the calpain-2-dependent cleavage of JPH2 in preclinical models of pressure overload and adrenergic stimulation has been described and also observed in ventricular samples from HF patients [
90]. Most interesting, however, is that contrary to the effects of JPH2NT, the blockade of nuclear localization of JPH2CT protected cardiomyocytes from isoproterenol-induced hypertrophy. Considering that these two fragments modulate cardiomyocyte growth in apparently opposite directions and that each one is generated by a specific calpain isoform, it can be speculated that the intracellular Ca
2+ concentration will determine the predominant JPH2 fragment. According to this hypothesis, JPH2CT, resulting from calpain-2, may have a preferential contribution in pathological conditions, while JPH2NT, resulting from the calpain-1 activity, in physiological or compensatory conditions. More recently, it has been suggested that calpain-2 can also cleave JPH2 at the same site as calpain-1, although with less efficacy [
135].
2. Calpain Contribution to the Progression of Pathologic Hypertrophy
In addition to their direct effect on the genesis of hypertrophy, calpains have also been proposed to participate in the progression of hypertrophy to HF.
2.1. Proteolysis of Myosin Light Chain Kinase
Evidence from studies conducted in a knockout myosin light chain kinase (MLCK) mouse model and a cardiac-specific overexpressing MLCK transgenic mouse model suggests an important role for cardiac myosin light chain (MLC) phosphorylation in the evolution of cardiac hypertrophy to HF [
136,
137]. Importantly, it has been described that β-adrenergic stimulation of neonatal cardiomyocytes and pressure overload produced by TAC induce a calpain-dependent proteolysis of MLCK [
138].
2.2. Mitochondrial Damage
Although traditionally considered cytoplasmic proteases, different studies have reported that calpains 1, 2, 4, and 10 are also found in mitochondria [
139,
140,
141]. Accumulating evidence, recently reviewed by Zhang et al. [
142], suggests that both cytosolic and mitochondrial calpain dysregulation may induce mitochondrial damage. Cardiac energy deprivation and increased ROS production resulting from mitochondrial dysfunction due to calpain overactivation during hypertrophy may promote the transition to decompensated hypertrophy and HF [
143].
Calpain overactivation was initially linked to the induction of the mitochondrial-dependent apoptotic program by activating the pro-apoptotic factors Bid [
144,
145] and AIF [
140]. However, the relevance of apoptotic cardiomyocyte death in the context of acute reperfusion injury and cardiac remodeling has been questioned due to the repression of the canonical caspase pathway in post-mitotic cardiomyocytes [
146,
147].
Instead, more recent studies propose that mitochondrial calpains contribute to the direct damage of the electron transporter chain (ETC) by targeting the NDUFS7 [
148] and ND6 [
141] subunits of complex I. Mitochondrial calpains have also been involved in the disruption of the mitochondrial FoF1 ATP synthase through the proteolysis of its ATP5A1 subunit [
149]. More recently, by using transgenic mice with calpain-1 upregulation restricted to cardiomyocyte mitochondria, the same group has demonstrated a causal association between calpain-mediated cleavage of ATP5A1 and ROS generation, mPTP opening, and cell death [
150].
Finally, mitochondrial calpains have been suggested to alter mitochondrial dynamics [
151,
152]. Mitofusin 2, which plays a central role in mitochondrial fusion, has been identified as a direct substrate of calpains [
153]. More recently, cardiac-specific downregulation of OPA1, a dynamin-related GTPase protein involved in mitochondrial fusion [
154] and in maintaining mitochondrial cristae structure [
155], has been associated with mitophagy inhibition and enhanced cardiomyocyte death in the setting of myocardial infarction [
156]. In a recent study, calpastatin overexpression in mice subjected to myocardial infarction prevented OPA1 degradation and improved mitochondrial fusion and mitophagy [
157]. However, whether calpain directly targets OPA1 or modulates OPA1 expression through an indirect mechanism remains to be elucidated. Furthermore, calpain inhibition has been shown to prevent beclin-1 cleavage, a key component of the autophagy pathway required to form autophagosomes, and improved mitophagy in isolated hearts subjected to transient ischemia [
148]. Altogether, these studies suggest that calpain overactivation negatively modulates mitophagy by acting at multiple levels.
3. Pharmacological Inhibition of Calpains
Despite the accumulating experimental evidence supporting the contribution of calpains to the development of hypertrophy, myocardial remodeling, and its progression to HF, no clinical trials have explored the pharmacological inhibition of calpains as a therapeutic strategy yet. The main reason for this is related to the limitations of most of the available calpain inhibitors, which involve low selectivity, limited membrane permeability, and reduced water solubility and metabolic stability [
52]. However, the research in the development of novel calpain inhibitors has been greatly benefited from the increasing evidence demonstrating the contribution of calpains to pathologic processes involved in adverse myocardial remodeling other than hypertrophy, including fibrosis and inflammation [
158], and non-cardiac pathologies, such as neurodegenerative disorders [
146], ophthalmic diseases [
159], myopathies [
160], and cancer [
161] (extensively discussed in previous reviews [
162]).
E-64 was the first calpain inhibitor used in rats to suggest the involvement of calpains in cardiac hypertrophy [
104]. E-64 and leupeptin constitute the first generation of calpain inhibitors, and their structure contains a peptidyl backbone and an electrophilic warhead that covalently interacts with the active site cysteine of calpain. However, although extensively used, these molecules show limited specificity for calpains and low membrane permeability [
163]. The pharmacological properties of leupeptin were improved by substituting its amino-terminal for a hydrophobic cap group [
163]. The inhibition of calpain activity using one of these synthetic leupeptin derivatives, MDL-28170 (calpain inhibitor III), has demonstrated efficacy against pathological hypertrophy and cardiac dysfunction in multiple rodent models of HF, including MI, TAC, and chronic isoproterenol infusion [
41]. However, these compounds show poor drug-like properties due to insufficient bioavailability and unfavorable pharmacokinetics, and, therefore, their progression into the clinic has been excluded. Advances in the design of new peptidomimetic calpain inhibitors have provided new molecules with improved water solubility and metabolic stability over previous inhibitors. Among them, and deriving from the benzoylalanine-derived ketoamide calpain inhibitor A-705253, A-953227 showed potent calpain inhibitory properties combined with high selectivity versus related cysteine protease cathepsins, other proteases, and receptors and was effective in reducing infarct size in an in vivo pig model of IR [
164]. However, the short effective half-life and low bioavailability caused by the instability against carbonyl reductases led to the development of a more metabolic stable derivative Alicapistat (ABT-957) [
165]. Alicapistat reached a phase I clinical study that analyzes its safety and pharmacological properties for the treatment of Alzheimer’s disease [
166]. However, although in preclinical models, alicapistat demonstrated efficacy with respect to the prevention of NMDA-induced neurodegeneration, it failed to induce any measurable hemodynamic effect in humans. This negative result was attributable to the use of an inadequate concentration and suggested a moderate inhibitory potency. The ketoamine derivative SNJ-1945, produced by Senju Pharmaceutical [
167,
168], shows an appropriate pharmacological profile, and its chronic oral administration was effective in preventing calpain activation and attenuating cardiomyocyte hypertrophy and cardiac dysfunction in a mouse model of transient LAD occlusion [
40]. More recently, these favorable effects of SNJ-1945 have been confirmed in a model of hypertrophy induced by chronic isoproterenol administration [
38]. Currently, a phase IIa clinical trial designed to test the efficacy and safety of the oral administration of SNJ-1945 in patients with non-arteritic retinal artery occlusion is in progress (jRTC2021190013).
The major limitation of calpain inhibitors is still their limited specificity for calpains over other cysteine proteases, mainly caused by the highly conserved active site among this type of proteases. A different approach aimed at increasing the specificity for calpains in the design of molecules that induce the allosteric inhibition of the enzyme by binding to other positions than the catalytic site. One of these allosteric inhibitors, PD150606, which is supposed to bind to the Ca
2+ binding site of calpain, was effective in attenuating the development of hypertrophy in isolated cardiomyocytes treated with isoproterenol [
169] or angiotensin II [
170]. It is important to mention that a PD150606 derivative, PD151746, is proposed to be more effective in inhibiting calpain-1 than calpain-2 [
171]. Considering that these two main calpain isoforms may display some differences in their substrate preference [
172] and biological function [
173,
174], this type of compound opens the door to the development of new isoform-selective inhibitors.
Calpains also seem to show differences in their regulation depending on their intracellular localization. It has been proposed that the activity of mitochondrial but not cytosolic calpains is regulated by its binding to chaperons (ERp57 for calpain-1 and Grp75 for calpain-2) [
175], and the use of peptides that blocks their interaction inhibits the mitochondrial activity of calpain-1 in a specific manner [
176]. Considering that mitochondria play a critical role in the development of hypertrophy [
177,
178] and that mitochondrial calpain-1 and/or calpain-2 increase in response to pathological stress associated with the development of HF [
150,
179], the potential therapeutic benefits of the compounds selectively addressed to mitochondrial calpains deserve further investigation.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23084103