Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Oligodendrocyte (OL) myelination is a critical process for the neuronal axon function in the central nervous system. OL myelination is critical to the vertebrate central nervous system (CNS) function. It supports not only the myelinating cell in the CNS but also provides metabolic and trophic support to the myelinated axon. The myelin sheath is essential insulation surrounding axons for conduction in the nervous system. Hypermyelination or hypomyelination interferes with saltatory nerve conduction, causing neurological disabilities.
- oligodendrocyte
- myelination
- proliferation
1. Introduction
OL progenitor cells (OPCs) come originally from neuroepithelial precursor cells and then proliferate and differentiate into premyelinating OLs, subsequently differentiating into myelinating OLs in the CNS (Figure 1), where an individual OL can myelinate up to 40 axonal segments [1][2][3][4]. Sequential generation of OPCs from specific germinal regions is reviewed by S. Mitew [5].
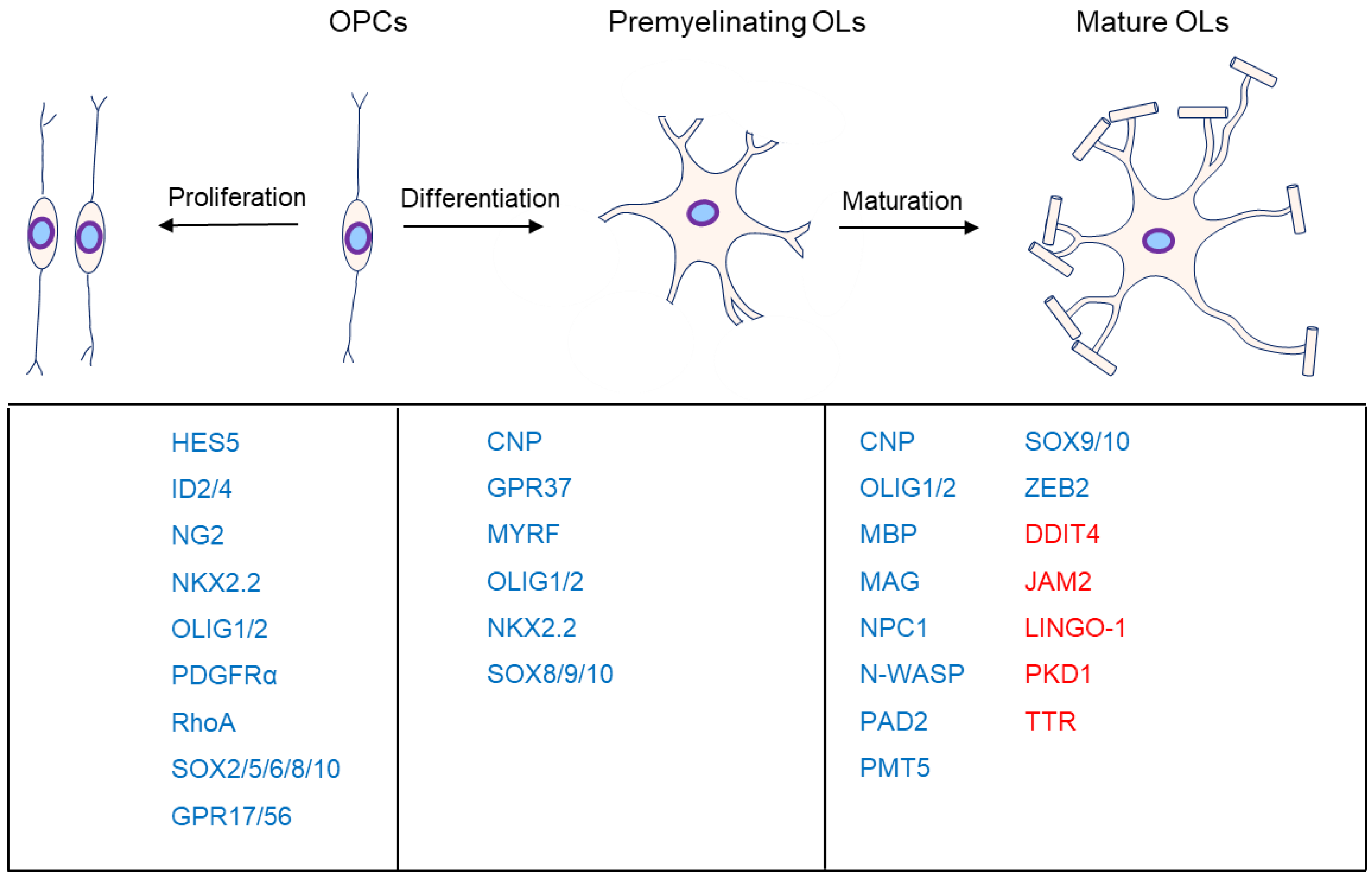
Figure 1. Major markers at the different stages of oligodendrocyte development. OL myelination is mainly divided into proliferation, differentiation, and maturation. The genes associated with the different stages are listed below them. Moreover, genes in blue are the positive markers in OPC proliferation, OL differentiation, or maturation. Specifically, genes in red are inhibitors in OL maturation.
2. OL Differentiation
OLs generated from OPCs are fundamental to myelin formation, and all myelination processes are composed of OPC proliferation, OL differentiation, and maturation. Although a supra-threshold axon diameter (>0.3 μm) is necessary for axonal myelination during optic nerve development, OL differentiation is independent of dynamic signals from the myelinated neuron [6]. Supra-threshold axon diameter is only an insufficient necessary factor.
Notably, the first territories to be occupied by OPCs are not necessarily the first regions to be myelinated [7], suggesting a potential regulation of other environmental factors. However, dynamic neuronal signals such as transcriptional changes or neuronal activity may also mediate physiological processes, such as OL differentiation and maturation [8]. The optic nerves are almost myelinated, but most brain regions are partially myelinated, even though the axon caliber is far more than the supra-threshold. Consequently, potential inhibitory factors or repulsive signals likely prevent the onset of myelination [9]. Conversely, it is also possible that attractive factors or signals exist to initiate the onset of myelination [6].
2.1. AKT-mTOR
AKT (AKT serine/threonine kinase 1), as an essential effector of PI3K (phosphatidylinositol-4,5-bisphosphate 3-kinase), promotes myelination in the CNS [10][11] through the mTOR (mechanistic target of rapamycin) pathway (Figure 2). Akt stimulates axonal wrapping and raises myelin thickness by the mTOR pathway. Moreover, maintaining AKT activation induces hypermyelination or demyelination [12]. Accordingly, sustaining the PI3K signaling pathway in OPCs results in gradual hypermyelination, leading to leukodystrophy [13]. Conversely, PTEN (phosphatase and tensin homolog) [14], as an inhibitor of the PI3K-Akt-mTOR signaling pathway, negatively regulates myelination, and DLG1 (discs large MAGUK scaffold protein 1, an interactor of PTEN) has a similar effect during myelination [13][15]. CNP (2,3-cyclic nucleotide 3-phosphodiesterase) controlling constitutive AKT activation boosts MBP staining and increases the corpus callosum size, with pronounced hypermyelination in small-caliber axons [12].
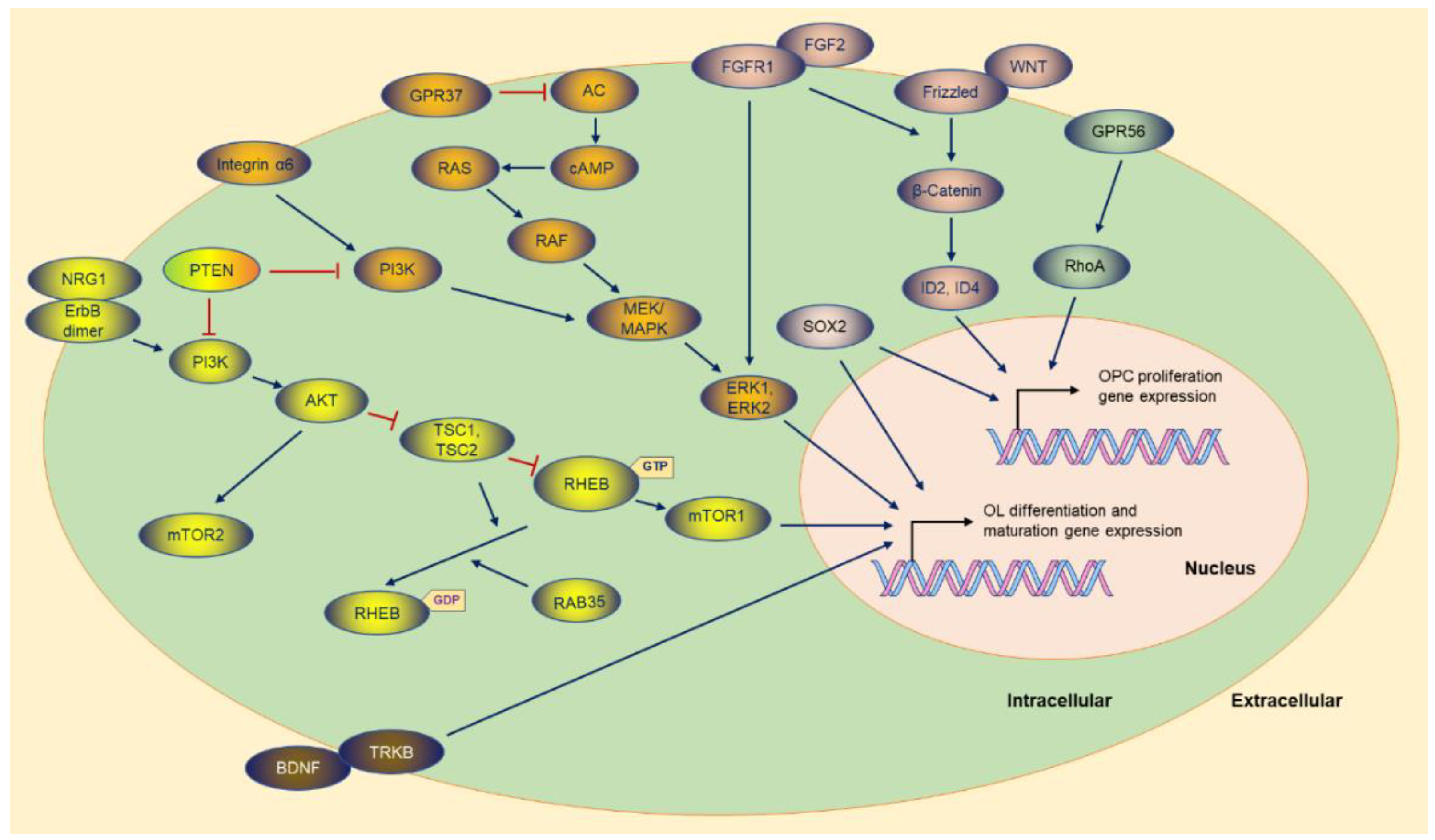 Figure 2. The molecules and the potential signaling pathway regulate the OPC proliferation and OL differentiation gene expression. Most of the molecules are exhibited, and the majority of the upstream molecules originate from the cytoplasmic membrane. The sign of blue arrows represents promoting, and the sign of red lines represents inhibiting. AC: Adenylate cyclase.
Figure 2. The molecules and the potential signaling pathway regulate the OPC proliferation and OL differentiation gene expression. Most of the molecules are exhibited, and the majority of the upstream molecules originate from the cytoplasmic membrane. The sign of blue arrows represents promoting, and the sign of red lines represents inhibiting. AC: Adenylate cyclase.mTOR, as the downstream of the PI3K-AKT signaling pathway, is the core element of two complexes (mTORC1 and mTORC2) with different functions in the complex [14][16]. Ablation of RAPTOR (regulatory-associated protein of MTOR complex 1), as an element of the mTORC1, leads to hypomyelination. Conversely, RICTOR (RPTOR independent companion of MTOR complex 2) deficiency does not affect myelination as an essential element of the mTORC2 [14].
Tuberous sclerosis protein 1 (TSC1) and TSC2 negatively regulate mTORC1 signaling [14][17][18], and downregulation of mTORC1 is essential for OPC differentiation and the subsequent myelination initiation. TSC2, as a member of GAP (GTPase-activating protein), constrains mTORC1 by stimulating the GTPase activity of RHEB (Ras homolog, mTORC1 binding), while RHEB binding to GTP causes the activation of mTORC1 [18]. Although TSC1 is not a GAP (RAS p21 protein activator 1), it stabilizes TSCs to maintain GTPase activity [14]. Thus, TSC1 or TSC2 ablation is supposed to strengthen RHEB-GTP stability, and consequently, abnormal activation of mTORC1 results in arrested differentiation in the early stage [16][19]. In some situations, mTORC1 hyperactivity causes distinctly delayed myelination and remyelination after injury [19]. Surprisingly, TSC1 deletion is detrimental to OL myelination [20][21].
mTORC1 activity also is downregulated by RAB35 (RAB35, member RAS oncogene family), a Ras-related GTPase controlling myelin growth through MTMR2 (myotubularin-related protein 2) and MTMR13 [22]. Accordingly, disruption of Rab35 causes hypermyelination via elevation of PI3P signaling and mTORC1 hyperactivation [22].
Some factors can switch the PI3K pathway to the MAPK pathway, such as Integrin α6 in OLs, a receptor for laminins in the neuronal axon. When the two types of molecules contact each other, myelin-forming OLs activate the switch in survival signaling dependence [23], which reverses neuregulin’s inhibition in OL differentiation, subsequently promoting the myelination process [23][24].
2.2. ERK1/2
The ERK1/2 advocates myelin wrapping during myelination and remyelination, and maintained OL ERK1/2 activation causes hypermyelination in the CNS [25] (Figure 2). ERK1/2 contributes to remyelination after the demyelination induced by lysophosphatidylcholine (LPC) injection into the corpus callosum [26]. MEK1 (MAPK kinase 1) is the upstream activator of ERK1/2 and displays declined expression in experimental autoimmune encephalomyelitis (EAE) induction [25]. Accordingly, maintained ERK1/2 activation upgrades myelin thickness after the LPC injection [27][28]. MEK inhibitors, such as PD0325901, AZD6244, AZD8330, CI-1040, and U0126 [29][30][31], significantly rectify OPC differentiation in a time- and dose-dependent manner, suggesting that regulation of the MAPK–ERK signaling pathway is sufficient to accelerate OL generation, facilitating myelin sheath formation [29].
2.3. GPR37
GPR37 is highly expressed in OLs and significantly intensified during OL differentiation and myelination (Figure 2). GPR37, also known as PAELR (Parkin-associated endothelin B-like receptor), has been identified as a substrate of parkin (an E3 ubiquitin ligase) [32]. Although the OPC number was not affected in Gpr37 null mice, GPR37 is regarded as an inhibitor of OL differentiation and myelination. Yang HJ et al. found that GPR37 restricts OL differentiation and hypermyelination by suppressing the cAMP-dependent Raf-MAPK-ERK1/2 cascade [33]. Gpr37 knockout leads to dramatically diminished MAG expression in the mouse brain. The mutant mice exhibit strikingly enlarged myelin loss during the cuprizone demyelination model without impacting the number of OPCs and OLs [34]. GPR37 suppresses activation of the cAMP-dependent Raf-MAPK-ERK1/2 cascade via inhibiting AC (adenylate cyclase) [33].
2.4. SOX10-MYRF
MYRF (myelin regulatory factor) is a membrane-bound transcriptional factor on the endoplasmic reticulum (ER). It forms homo-trimers in the ER [35] and then undergoes self-cleavage via the intrinsic peptidase [36][37][38]. TMEM98 (transmembrane protein 98) can block MYRF self-cleavage in vitro and in vivo [39][40]. Following the self-proteolysis, the homo-trimer of MYRF N-terminal fragments is translocated to the nucleus and binds the motif to activate the transcription of myelin genes [36][37][41][42][43][44][45][46].
MYRF is required to initiate and maintain myelination [5][47] (Figure 2). Myrf knockout mice sustain the premyelinating stage, leading to myelination failure and postnatal death [48][49], a similar phenotype to that in Olig1 null mice [50] and Sox10 mutant Zebrafish [51]. Loss of Myrf in OPCs does not alter OPC proliferation and recruitment after demyelination but impairs remyelination because of diminishing OL differentiation [41][52]. In addition, OLs deriving from OPCs with Myrf deletion produce few myelin proteins in response to demyelination [41]. Not surprisingly, conditioned knockout Myrf in mature OLs leads to a dramatic downregulation of myelin gene expression and impairment of myelin sheaths [47][52].
Myrf expression is enormously intensified during the initiation of OL differentiation, and SOX10 acts as a Myrf gene enhancer [53]. SOX10 binds the first intron of Myrf, which is also the region regulated by OLIG2 [54]. Once induced by SOX10, MYRF redirects SOX10 to myelin gene expression [46]. Therefore, the feedback and forward regulatory loops compose an essential molecular circuit for OL differentiation and maturation. Surprisingly, despite the importance of MYRF in OL differentiation and maturation, most of the genetic mutations of Myrf do not cause apparent myelin-related human diseases [55][56][57][58][59].
2.5. NRG1
NRG1 (Neuregulin-1) promotes OPC migration, proliferation, and differentiation into OLs [60][61][62], and NRG1/ErbB (Erb-b2 receptor tyrosine kinase) signaling (Figure 2) is required for OL survival and maturation [63][64][65].
NRG1 has more than 30 splice isoforms, sharing an EGF-like function domain to activate the receptors, ErbB2/ErbB3 heterodimer, or ErbB4 homodimer [66][67][68]. Immature NRG1 is a transmembrane protein that releases soluble N-terminal moieties containing the EGF-like domain after proteolytic processing [68]. Soluble NRG1 is a mitogen for OLs, provides an axonal signal for OL survival and increases myelination [69]. Inhibiting its receptors reversed the positive effects of the administration of soluble NRG1 [60]. BACE1 (Beta-site APP-cleaving enzyme 1) cleaves NRG1 type Ⅰ and type Ⅲ. Bace1 deficiency leads to hypomyelination and impairs remyelination [64].
NRG1, acting through ErbB, is an important regulators in OPC proliferation and OL differentiation during development [70]. NRG1-ErbB modulates myelin-related gene expression depending on the PI3K-AKT-mTOR pathway [67][71][72]. Recently, Ding Z et al. found that NRG1 can convert astrocytes into OL lineage cells via the PI3K-AKT-mTOR signaling activation and eventually improves remyelination [73]. Increasing AKT activity of the OLs in Bace1 null mice is sufficient to normalize myelination without inducing hypermyelination [74].
Nrg1 type Ⅲ null mice died at birth owing to the deficiency of functional neuromuscular junction, which also hampers the analysis of NRG1 in the CNS [75][76]. Although OLs normally differentiate when cocultured with Nrg1 type Ⅲ null dorsal root ganglia, the myelination of dorsal root ganglia is impaired in the Nrg1 type Ⅲ deficient mice [77]. Intriguingly, heterozygous Nrg1 type Ⅲ mutant mice exhibit hypomyelination in the brain, but myelination in the spinal cord and the optic nerve is normal [77]. However, others have not found CNS hypomyelination in heterozygous Nrg1 type Ⅲ mutant or Nrg1 null mice [78]. Surprisingly, mice overexpressing Nrg1 type Ⅲ have hypomyelinated axons in the CNS [78]. Nrg1 type Ⅲ overexpression in the spinal cord improves motor function and increases motor neuron survival in mice with amyotrophic lateral sclerosis [79].
2.6. BDNF
CNS myelinating cells develop from slowly dividing adult progenitor cell OPCs through a premyelinating OL stage before maturing into myelinating OLs. Bdnf (brain-derived neurotrophic factor) heterozygous mice display hindered myelination in the CNS [80] (Figure 2). BDNF functions via two different classes of transmembrane receptors: TRKB (tropomyosin-related kinase receptor B) and p75NTR (p75 neurotrophin receptor). Maintaining BDNF enhances myelination, but it cannot be attenuated in the p75Ntr knockout mice [80]. Therefore, p75NTR is not necessary for promoting myelination. Conversely, BDNF cannot rescue the impeding myelination induced by the impairment of TrkB signaling [80].
2.7. GlcNAc
Recently, Michael et al. [81] found that GlcNAc (N-acetylglucosamine) is essential in triggering OL differentiation. N-glycan branching and GlcNAc support OPC differentiation into OLs via suppressing PDGF-α. Furthermore, primary myelination in newborn pups is strengthened when the lactating mice are supplemented with oral GlcNAc. Conversely, by blocking N-glycan branching, primary myelination is impeded. Intriguingly, oral GlcNAc protects neuronal axons from damage in the cuprizone-induced demyelination mouse model via enhancing myelin reparation [81].
2.8. OLIG2
OLIG2 is regarded as a marker of OL family cells, although it is also expressed during development in motoneurons, subgroups of astrocyte precursors, and Purkinje cell precursors [82][83][84][85][86]. OLIG2 acts as a binding upstream enhancer to induce the expression of target genes such as Nkx2.2 (NK2 homeobox 2) and Sox10. Conditional knockout of Olig2 in embryonic neural stem cells confines OL differentiation without the alteration of OL specification, resulting in hypomyelination [87].
OLIG2 is critical to OPC specification and OL differentiation. When cortical OPCs are conditional knockout Olig2, the OPCs are transformed into astrocytes [88]. OPCs are completely absent from most regions of the CNS in Olig2 knockout mice [89][90][91][92]. However, OPCs are still present in dramatically reduced numbers in the forebrain and hindbrain of Olig2 knockout mice [89][93]. Conditioned knockout of Olig2 in OPCs leads to hypomyelination because of the limitation of OL differentiation, while conditional knockout of Olig2 in immature OLs accelerates OL myelination by facilitating maturation [93].
Though Olig2 is closely related to Olig1 [94], Olig1 provides little compensation for Olig2 loss [82]. OLIG2 forms a homodimer or a heterodimer to induce OPC specification [83]. Moreover, OLIG2 exhibits versatile functions in OPCs and OLs via post-translational modification, which was reviewed thoroughly by H Li and WD Richardson [86].
2.9. PDE
PDEs (phosphodiesterases) have been implicated in OL maturation and myelination in the CNS (Figure 2). Inhibitors of PDEs, such as PDE1 inhibitor vinpocetine [95] and PDE5 inhibitor sildenafil [96], not only spoil inflammation but also exert a negative impact on the CNS OL differentiation processes, including diminishing myelin gene expression and enhancing myelination negative transcriptional regulators (such as ID2 and ID4) [96].
3. OL Maturation
3.1. DDIT4
DDIT4 (DNA damage inducible transcript 4, also known as REDD1/Dig2/RTP801) is a negative regulator of myelination [15]. DDIT4 maximum expression is in line with the peak activity of AKT and DLG1. Moreover, Ddit4 deficiency provokes hypermyelination by enhancing mTOR activation and enlarged myelin thickness. Intriguingly, Ddit4-deficient mice do not present myelin out-folding (depending on PIP3) or macula (depending on AKT) [15].
3.2. JAM2
Neuronal JAM2 (junction adhesion molecule 2) is sufficient and necessary in the somatodendritic membrane to inhibit OL myelination in the neuronal cell body [9][97], while Galectin-4 is an inhibitor in the axons [98]. Galectin-4 is specifically sorted into segmental domains along the axon membrane, and OLs do not deposit myelin on Galectin-4 covered surfaces, leading to long unmyelinated axon segments [98]. After the JAM2 extracellular portion is fused to the immunoglobulin Fc region, the formed JAM2-Fc is added into cultured OLs, with higher JAM2-Fc binding to MBP+ myelinating OLs than to OPCs, suggesting that the JAM2 receptor is upregulated on the surface during OL differentiation [9][97]. Furthermore, soluble JAM2-Fc arrests myelin formation in cultured OLs from wild-type mice [9][97]. Intriguingly, recent studies have found that JAM2 is also associated with primary familial brain calcification, an uncommon degenerative neurological disease due to abnormal calcium phosphate deposits in the brain [99][100]. In Jam2 null mice, the neuronal soma is sheathed, and contactin-associated protein, which typically localizes only with the paranodal structures of the Nodes of Ranvier, clusters on the neuronal somatic member [9].
3.3. PKD1
PKD1 (protein kinase D1), a serine/threonine kinase belonging to the calcium/calmodulin-dependent kinase family, is implicated in OL maturation, except for its role in tumor progression [101][102]. Pkd1 homozygous deficiency is lethal to the mutant mice, and Pkd1 heterozygous mutant mice exhibit quickly inducible epilepsy and hypermyelination, supporting the finding of the epilepsy patient [103]. In addition, PKD1 can elevate functional synapse formation by enhancing N-cadherin’s stability in an activity-dependent manner [104].
3.4. TTR
TTR (Transthyretin) binds and transfers thyroid hormones in cerebrospinal fluid and blood. Ttr mutation is related to familial amyloid polyneuropathy, a neurodegenerative disorder with TTR deposition in the peripheral nervous system [105][106]. However, Ttr null mice exhibit hypermyelination, magnified OL density in the corpus callosum, and anterior commissure during postnatal development [107]. Moreover, Ttr deficiency magnifies OPC migration and proliferation with debased apoptosis [107]. Intriguingly, TTR is expressed in OPCs, and boosting the pAKT level in OLs may be the mechanism of hypermyelination [107]. During remyelination in the adult mouse corpus callosum, Ttr null mice exhibit an expedited remyelination rate, preferentially remyelinating small axons [108]. Moreover, Ttr null mice display thicker myelin than wild-type mice [108].
3.5. LINGO-1
LINGO-1 (leucine-rich repeat and Ig-like domain-containing Nogo receptor interacting protein 1), a transmembrane protein, is expressed explicitly in OLs and neurons, serving as a potent negative modulation of axonal myelination and regeneration in the CNS [3][109]. Downregulating LINGO-1 functions [3], such as Lingo-1 RNAi [110], sh-RNA [109][111], anti-LINGO-1 antibody [112][113][114][115][116], dominant-negative LINGO-1, or soluble LINGO-1-Fc [117][118], improves OL differentiation and myelination, accompanied by prolonged process length and augment branching, and downregulated RhoA [116][119] activity is the potential mechanism [3]. Conversely, Lingo-1 overexpression results in RhoA activation, negatively regulating OL differentiation and myelination [3]. Neuronal LINGO-1 is a critical component of the Nogo receptor complex, restricting axonal growth via RhoA. The Nogo receptor is absent in OLs, and consequently, LINGO-1 prevents OL myelination through intercellular interactions with self-association in the trans [118] or cytoplasmic gelsolin signaling pathway [114].
3.6. N-WASP
N-WASP (Neural Wiskott–Aldrich syndrome protein) is essential for myelin wrapping in Schwan cell and OL myelination [120][121][122][123]. Conditional knockout N-Wasp continues to ensheathe the onset myelinating axons but fails to extend circumferentially to elaborate myelin, and the affected mice demonstrate apparent motor deficits without progress [122]. In N-Wasp-deficient nerves, most cells arrest at the premyelinating stage and subsequently fail to myelinate, with occasional misfolding myelin forming unusually short internodes and thin myelin sheaths [121]. Strikingly, N-Wasp deficiency leads to hypomyelination and induces remarkably focal hypermyelination, representing long myelin out-folds enclosing neuronal cell bodies and unmyelinated axons [120].
3.7. PRMT5
PRMT5 (protein arginine methyltransferase 5) is a histone arginine methyltransferase catalyzing histone H4R3 methylation. Although suppression or knockout of Prmt5 does not affect OPC proliferation, it attenuates OPC survival and differentiation leading to hypomyelination [124]. Its potential mechanism is heightened nuclear acetylation of H4K5 following histone H4R3 methylation reduction, which can be rescued via bridling histone acetyltransferases [124].
3.8. ZEB2
ZEB2 (zinc finger E-box binding homeobox 2, also known as Zfhx1b and Sip1), a transcription factor, contributes to many essential neurodevelopmental processes [125][126][127][128]. ZEB2 heterozygous mutation in humans leads to Mowat–Wilson syndrome [125][128]. Zeb2 knockout mice have severe impairment of myelination, failing to express myelin genes, and ZEB2 may be one of the direct transcriptional targets of Olig2 [129][130]. Moreover, ZEB2 controls the onset of Schwann cell differentiation by recruiting HDAC1/2 and nucleosome remodeling and deacetylase complex co-repressor complexes in mice [127].
3.9. PAD2
Citrullination, a modification converting peptidyl-arginine residues to peptidyl-citrulline, has been associated with the etiology of several diseases, including inflammation in the CNS [131]. Citrullination by PAD2 (peptidyl-arginine deiminase 2) [132] contributes to OL differentiation and myelination by modifying myelin and chromatin-related proteins [133]. In Pad2 transgenic mice, homozygous mice exhibit thinner myelin and more severe focal demyelination than heterozygous mice [134]. Overexpression of Pad2 increases levels of TNF-α (tumor necrosis factor-α), TNF-α induces predominantly cytosolic PAD4 translocation into the nucleus, and high citrullination of histones by PAD4 causes irreversible changes to OLs, which may contribute to apoptosis [134][135]. Moreover, citrullination of MBP by PAD2 leads to reduced interaction of the arginine residues with negatively charged lipids, forming incompact sheaths lacking an attraction between the MBP and lipids, and consequently, the myelin becomes unstable or remains immature [136].
3.10. NPC1
CNS hypomyelination is one of the pathological characteristics of Niemann–Pick Type C disease (NPC), a rare childhood-onset neurodegenerative disorder due to mutations of NPC1 (NPC intracellular cholesterol transporter 1) or NPC2 [137]. Npc1-deficient mice display hypomyelination and delayed myelination caused by hampered OL maturation [137][138]. NPC patients suffer abnormally swollen axons and intracellular lipid accumulation [139]. A deficiency of NPC1, a transmembrane protein essential for mobilizing cholesterol from late endosomes and lysosomes, in neurons alone does not affect the density of OPCs but results in an arrest of OL maturation [139]. Deletion of Npc1 in OLs leads to a delay rather than a block of myelination [139]. Npc1 is also required for CNS myelin maintenance because OL-conditioned knockout Npc1 in aged mice causes late-stage myelin loss, followed by secondary Purkinje neuron degeneration [139].
This entry is adapted from the peer-reviewed paper 10.3390/cimb44050149
References
- van Tilborg, E.; de Theije, C.G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238.
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782.
- Mi, S.; Miller, R.H.; Lee, X.; Scott, M.L.; Shulag-Morskaya, S.; Shao, Z.; Chang, J.; Thill, G.; Levesque, M.; Zhang, M.; et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 2005, 8, 745–751.
- Tian, C.; Zou, S.; Hu, B. Extraocular Source of Oligodendrocytes Contribute to RetinalMyelination and Optokinetic Responses in Zebrafish. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2129–2138.
- Mitew, S.; Hay, C.M.; Peckham, H.; Xiao, J.; Koenning, M.; Emery, B. Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience 2014, 276, 29–47.
- Mayoral, S.R.; Etxeberria, A.; Shen, Y.-A.A.; Chan, J.R. Initiation of CNS Myelination in the Optic Nerve Is Dependent on Axon Caliber. Cell Rep. 2018, 25, 544–550.e543.
- Sabatelli, M.; Mignogna, T.; Lippi, G.; Servidei, S.; Manfredi, G.; Ricci, E.; Bertini, E.; Monaco, M.L.; Tonali, P. Autosomal recessive hypermyelinating neuropathy. Acta Neuropathol. 1995, 87, 337–342.
- Mitew, S.; Gobius, I.; Fenlon, L.R.; McDougall, S.J.; Hawkes, D.; Xing, Y.L.L.; Bujalka, H.; Gundlach, A.L.; Richards, L.J.; Kilpatrick, T.J.; et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. Commun. 2018, 22, 306.
- Redmond, S.A.; Mei, F.; Eshed-Eisenbach, Y.; Osso, L.A.; Leshkowitz, D.; Shen, Y.A.; Kay, J.N.; Aurrand-Lions, M.; Lyons, D.A.; Peles, E.; et al. Somatodendritic Expression of JAM2 Inhibits Oligodendrocyte Myelination. Neuron 2016, 91, 824–836.
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 6860–6870.
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.; Kidd, G.; Avila, R.L.; Kirschner, D.A.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 7174–7183.
- Domenech-Estevez, E.; Baloui, H.; Meng, X.; Zhang, Y.; Deinhardt, K.; Dupree, J.L.; Einheber, S.; Chrast, R.; Salzer, J.L. Akt Regulates Axon Wrapping and Myelin Sheath Thickness in the PNS. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 4506–4521.
- Maire, C.L.; Ramkissoon, S.; Hayashi, M.; Haidar, S.; Ramkissoon, L.; DiTomaso, E.; Ligon, K.L. Pten loss in Olig2 expressing neural progenitor cells and oligodendrocytes leads to interneuron dysplasia and leukodystrophy. Stem Cells 2014, 32, 313–326.
- Beirowski, B.; Wong, K.M.; Babetto, E.; Milbrandt, J. mTORC1 promotes proliferation of immature Schwann cells and myelin growth of differentiated Schwann cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4261–E4270.
- Noseda, R.; Belin, S.; Piguet, F.; Vaccari, I.; Scarlino, S.; Brambilla, P.; Martinelli Boneschi, F.; Feltri, M.L.; Wrabetz, L.; Quattrini, A.; et al. DDIT4/REDD1/RTP801 is a novel negative regulator of Schwann cell myelination. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 15295–15305.
- Figlia, G.; Norrmen, C.; Pereira, J.A.; Gerber, D.; Suter, U. Dual function of the PI3K-Akt-mTORC1 axis in myelination of the peripheral nervous system. eLife 2017, 6, e29241.
- Kennedy, B.K.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003.
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293.
- Figlia, G.; Gerber, D.; Suter, U. Myelination and mTOR. Glia 2018, 66, 693–707.
- Jiang, M.; Liu, L.; He, X.; Wang, H.; Lin, W.; Wang, H.; Yoon, S.O.; Wood, T.L.; Lu, Q.R. Regulation of PERK-eIF2alpha signalling by tuberous sclerosis complex-1 controls homoeostasis and survival of myelinating oligodendrocytes. Nat. Commun. 2016, 7, 12185.
- Lebrun-Julien, F.; Bachmann, L.; Norrmen, C.; Trotzmuller, M.; Kofeler, H.; Ruegg, M.A.; Hall, M.N.; Suter, U. Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 8432–8448.
- Sawade, L.; Grandi, F.; Mignanelli, M.; Patino-Lopez, G.; Klinkert, K.; Langa-Vives, F.; Di Guardo, R.; Echard, A.; Bolino, A.; Haucke, V. Rab35-regulated lipid turnover by myotubularins represses mTORC1 activity and controls myelin growth. Nat. Commun. 2020, 11, 2835.
- Colognato, H.; Nishiyama, A. Introduction to the Special Issue on The oligodendrocyte niche in development and repair. Neurosci. Lett. 2020, 730, 134957.
- Baron, W.; Colognato, H.; ffrench-Constant, C. Integrin-growth factor interactions as regulators of oligodendroglial development and function. Glia 2005, 49, 467–479.
- Jeffries, M.A.; Obr, A.E.; Urbanek, K.; Fyffe-Maricich, S.L.; Wood, T.L. Cnp Promoter-Driven Sustained ERK1/2 Activation Increases B-Cell Activation and Suppresses Experimental Autoimmune Encephalomyelitis. ASN Neuro 2020, 12, 1–18.
- Michel, K.; Zhao, T.; Karl, M.; Lewis, K.; Fyffe-Maricich, S.L. Translational control of myelin basic protein expression by ERK2 MAP kinase regulates timely remyelination in the adult brain. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 7850–7865.
- Ishii, A.; Furusho, M.; Bansal, R. Sustained activation of ERK1/2 MAPK in oligodendrocytes and schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 175–186.
- Gaesser, J.M.; Fyffe-Maricich, S.L. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp. Neurol. 2016, 283, 501–511.
- Suo, N.; Guo, Y.E.; He, B.; Gu, H.; Xie, X. Inhibition of MAPK/ERK pathway promotes oligodendrocytes generation and recovery of demyelinating diseases. Glia 2019, 67, 1320–1332.
- Furqan, M.; Akinleye, A.; Mukhi, N.; Mittal, V.; Chen, Y.; Liu, D. STAT inhibitors for cancer therapy. J. Hematol. Oncol. 2013, 6, 90.
- Planz, O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antivir. Res. 2013, 98, 457–468.
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An Unfolded Putative Transmembrane Polypeptide, which Can Lead to Endoplasmic Reticulum Stress, Is a Substrate of Parkin. Cell 2001, 105, 891–902.
- Yang, H.J.; Vainshtein, A.; Maik-Rachline, G.; Peles, E. G protein-coupled receptor 37 is a negative regulator of oligodendrocyte differentiation and myelination. Nat. Commun. 2016, 7, 10884.
- Smith, B.M.; Giddens, M.M.; Neil, J.; Owino, S.; Nguyen, T.T.; Duong, D.; Li, F.; Hall, R.A. Mice lacking Gpr37 exhibit decreased expression of the myelin-associated glycoprotein MAG and increased susceptibility to demyelination. Neuroscience 2017, 358, 49–57.
- Kim, D.; Choi, J.O.; Fan, C.; Shearer, R.S.; Sharif, M.; Busch, P.; Park, Y. Homo-trimerization is essential for the transcription factor function of Myrf for oligodendrocyte differentiation. Nucleic Acids Res. 2017, 45, 5112–5125.
- ffrench-Constant, C.; Bujalka, H.; Koenning, M.; Jackson, S.; Perreau, V.M.; Pope, B.; Hay, C.M.; Mitew, S.; Hill, A.F.; Lu, Q.R.; et al. MYRF Is a Membrane-Associated Transcription Factor That Autoproteolytically Cleaves to Directly Activate Myelin Genes. PLoS Biol. 2013, 11, e1001625.
- Li, Z.; Park, Y.; Marcotte, E.M. A Bacteriophage tailspike domain promotes self-cleavage of a human membrane-bound transcription factor, the myelin regulatory factor MYRF. PLoS Biol. 2013, 11, e1001624.
- Wu, P.; Zhen, X.; Li, B.; Yu, Q.; Huang, X.; Shi, N. Crystal structure of the MyRF ICA domain with its upstream beta-helical stalk reveals the molecular mechanisms underlying its trimerization and self-cleavage. Int. J. Biol. Sci. 2021, 17, 2931–2943.
- Huang, H.; Teng, P.; Du, J.; Meng, J.; Hu, X.; Tang, T.; Zhang, Z.; Qi, Y.B.; Qiu, M. Interactive Repression of MYRF Self-Cleavage and Activity in Oligodendrocyte Differentiation by TMEM98 Protein. J. Neurosci. 2018, 38, 9829–9839.
- Cross, S.H.; McKie, L.; Hurd, T.W.; Riley, S.; Wills, J.; Barnard, A.R.; Young, F.; MacLaren, R.E.; Jackson, I.J. The nanophthalmos protein TMEM98 inhibits MYRF self-cleavage and is required for eye size specification. PLoS Genet. 2020, 16, e1008583.
- Duncan, G.J.; Plemel, J.R.; Assinck, P.; Manesh, S.B.; Muir, F.G.W.; Hirata, R.; Berson, M.; Liu, J.; Wegner, M.; Emery, B.; et al. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 403–422.
- Zhen, X.; Li, B.; Hu, F.; Yan, S.; Meloni, G.; Li, H.; Shi, N. Crystal structure of the DNA-binding domain of Myelin-gene Regulatory Factor. Sci. Rep. 2017, 7, 3696.
- Chen, B.; Zhu, Y.; Ye, S.; Zhang, R. Structure of the DNA-binding domain of human myelin-gene regulatory factor reveals its potential protein-DNA recognition mode. J. Struct. Biol. 2018, 203, 170–178.
- Fan, C.; An, H.; Sharif, M.; Kim, D.; Park, Y. Functional mechanisms of MYRF DNA-binding domain mutations implicated in birth defects. J. Biol. Chem. 2021, 296, 100612.
- Choi, J.O.; Fan, C.; Kim, D.; Sharif, M.; An, H.; Park, Y. Elucidating the transactivation domain of the pleiotropic transcription factor Myrf. Sci. Rep. 2018, 8, 13075.
- Aprato, J.; Sock, E.; Weider, M.; Elsesser, O.; Frob, F.; Wegner, M. Myrf guides target gene selection of transcription factor Sox10 during oligodendroglial development. Nucleic Acids Res. 2020, 48, 1254–1270.
- Koenning, M.; Jackson, S.; Hay, C.M.; Faux, C.; Kilpatrick, T.J.; Willingham, M.; Emery, B. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 12528–12542.
- Emery, B.; Lu, Q.R. Transcriptional and Epigenetic Regulation of Oligodendrocyte Development and Myelination in the Central Nervous System. Cold Spring Harb. Perspect. Biol. 2015, 7, e020461.
- Emery, B.; Agalliu, D.; Cahoy, J.D.; Watkins, T.A.; Dugas, J.C.; Mulinyawe, S.B.; Ibrahim, A.; Ligon, K.L.; Rowitch, D.H.; Barres, B.A. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 2009, 138, 172–185.
- Xin, M.; Yue, T.; Ma, Z.; Wu, F.F.; Gow, A.; Lu, Q.R. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1354–1365.
- Takada, N.; Kucenas, S.; Appel, B. Sox10 is necessary for oligodendrocyte survival following axon wrapping. Glia 2010, 58, 996–1006.
- McKenzie, I.A.; Ohayon, D.; Li, H.; de Faria, J.P.; Emery, B.; Tohyama, K.; Richardson, W.D. Motor skill learning requires active central myelination. Science 2014, 346, 318–322.
- Hornig, J.; Frob, F.; Vogl, M.R.; Hermans-Borgmeyer, I.; Tamm, E.R.; Wegner, M. The transcription factors Sox10 and Myrf define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet. 2013, 9, e1003907.
- Yu, Y.; Chen, Y.; Kim, B.; Wang, H.; Zhao, C.; He, X.; Liu, L.; Liu, W.; Wu, L.M.; Mao, M.; et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 2013, 152, 248–261.
- Kurahashi, H.; Azuma, Y.; Masuda, A.; Okuno, T.; Nakahara, E.; Imamura, T.; Saitoh, M.; Mizuguchi, M.; Shimizu, T.; Ohno, K.; et al. MYRF is associated with encephalopathy with reversible myelin vacuolization. Ann. Neurol. 2018, 83, 98–106.
- Rossetti, L.Z.; Glinton, K.; Yuan, B.; Liu, P.; Pillai, N.; Mizerik, E.; Magoulas, P.; Rosenfeld, J.A.; Karaviti, L.; Sutton, V.R.; et al. Review of the phenotypic spectrum associated with haploinsufficiency of MYRF. Am. J. Med. Genetics. Part A 2019, 179, 1376–1382.
- Garnai, S.J.; Brinkmeier, M.L.; Emery, B.; Aleman, T.S.; Pyle, L.C.; Veleva-Rotse, B.; Sisk, R.A.; Rozsa, F.W.; Ozel, A.B.; Li, J.Z.; et al. Variants in myelin regulatory factor (MYRF) cause autosomal dominant and syndromic nanophthalmos in humans and retinal degeneration in mice. PLoS Genet. 2019, 15, e1008130.
- Yu, X.; Sun, N.; Yang, X.; Zhao, Z.; Zhang, J.; Zhang, M.; Zhang, D.; Ge, J.; Fan, Z. Myelin regulatory factor deficiency is associated with the retinal photoreceptor defects in mice. Vis. Neurosci. 2021, 38, E005.
- Huang, H.; Zhou, F.; Zhou, S.; Qiu, M. MYRF: A Mysterious Membrane-Bound Transcription Factor Involved in Myelin Development and Human Diseases. Neurosci. Bull. 2021, 37, 881–884.
- Gauthier, M.K.; Kosciuczyk, K.; Tapley, L.; Karimi-Abdolrezaee, S. Dysregulation of the neuregulin-1-ErbB network modulates endogenous oligodendrocyte differentiation and preservation after spinal cord injury. Eur. J. Neurosci. 2013, 38, 2693–2715.
- Canoll, P.D.; Musacchio, J.M.; Hardy, R.; Reynolds, R.; Marchionni, M.A.; Salzer, J.L. GGF/Neuregulin Is a Neuronal Signal That Promotes the Proliferation and Survival and Inhibits the Differentiation of Oligodendrocyte Progenitors. Neuron 1996, 17, 229–243.
- Ortega, M.C.; Bribian, A.; Peregrin, S.; Gil, M.T.; Marin, O.; de Castro, F. Neuregulin-1/ErbB4 signaling controls the migration of oligodendrocyte precursor cells during development. Exp. Neurol. 2012, 235, 610–620.
- Kataria, H.; Alizadeh, A.; Shahriary, G.M.; Saboktakin Rizi, S.; Henrie, R.; Santhosh, K.T.; Thliveris, J.A.; Karimi-Abdolrezaee, S. Neuregulin-1 promotes remyelination and fosters a pro-regenerative inflammatory response in focal demyelinating lesions of the spinal cord. Glia 2018, 66, 538–561.
- Hu, X.; Hicks, C.W.; He, W.; Wong, P.; Macklin, W.B.; Trapp, B.D.; Yan, R. Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 2006, 9, 1520–1525.
- Makinodan, M.; Rosen, K.M.; Ito, S.; Corfas, G. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 2012, 337, 1357–1360.
- Mei, L.; Xiong, W.-C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 2008, 9, 437–452.
- Luo, X.; Prior, M.; He, W.; Hu, X.; Tang, X.; Shen, W.; Yadav, S.; Kiryu-Seo, S.; Miller, R.; Trapp, B.D.; et al. Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J. Biol. Chem. 2011, 286, 23967–23974.
- Mei, L.; Nave, K.A. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 2014, 83, 27–49.
- Wang, Z.; Colognato, H.; Ffrench-Constant, C. Contrasting effects of mitogenic growth factors on myelination in neuron-oligodendrocyte co-cultures. Glia 2007, 55, 537–545.
- Kataria, H.; Karimi-Abdolrezaee, S. Neuregulin-1: A novel regulator of glial response in spinal cord injury. Neural Regen. Res. 2017, 12, 1616–1617.
- Goebbels, S.; Oltrogge, J.H.; Kemper, R.; Heilmann, I.; Bormuth, I.; Wolfer, S.; Wichert, S.P.; Mobius, W.; Liu, X.; Lappe-Siefke, C.; et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 8953–8964.
- Nave, K.A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252.
- Ding, Z.; Dai, C.; Zhong, L.; Liu, R.; Gao, W.; Zhang, H.; Yin, Z. Neuregulin-1 converts reactive astrocytes toward oligodendrocyte lineage cells via upregulating the PI3K-AKT-mTOR pathway to repair spinal cord injury. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 134, 111168.
- Hu, X.; Schlanger, R.; He, W.; Macklin, W.B.; Yan, R. Reversing hypomyelination in BACE1-null mice with Akt-DD overexpression. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1868–1873.
- Wolpowitz, D.; Mason, T.B.A.; Dietrich, P.; Mendelsohn, M.; Talmage, D.A.; Role, L.W. Cysteine-Rich Domain Isoforms of the Neuregulin-1 Gene Are Required for Maintenance of Peripheral Synapses. Neuron 2000, 25, 79–91.
- Adlkofer, K.; Lai, C. Role of Neuregulins in Glial Cell Development. Glia 2000, 29, 104–111.
- Taveggia, C.; Thaker, P.; Petrylak, A.; Caporaso, G.L.; Toews, A.; Falls, D.L.; Einheber, S.; Salzer, J.L. Type III neuregulin-1 promotes oligodendrocyte myelination. Glia 2008, 56, 284–293.
- Brinkmann, B.G.; Agarwal, A.; Sereda, M.W.; Garratt, A.N.; Muller, T.; Wende, H.; Stassart, R.M.; Nawaz, S.; Humml, C.; Velanac, V.; et al. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 2008, 59, 581–595.
- Modol-Caballero, G.; Herrando-Grabulosa, M.; Verdes, S.; Garcia-Lareu, B.; Hernandez, N.; Francos-Quijorna, I.; Lopez-Vales, R.; Bosch, A.; Navarro, X. Gene Therapy Overexpressing Neuregulin 1 Type I in Combination With Neuregulin 1 Type III Promotes Functional Improvement in the SOD1(G93A) ALS Mice. Front. Neurol. 2021, 12, 693309.
- Xiao, J.; Wong, A.W.; Willingham, M.M.; van den Buuse, M.; Kilpatrick, T.J.; Murray, S.S. Brain-derived neurotrophic factor promotes central nervous system myelination via a direct effect upon oligodendrocytes. Neurosignals 2010, 18, 186–202.
- Sy, M.; Brandt, A.U.; Lee, S.-U.; Newton, B.L.; Pawling, J.; Golzar, A.; Rahman, A.M.A.; Yu, Z.; Cooper, G.; Scheel, M.; et al. N-acetylglucosamine drives myelination by triggering oligodendrocyte precursor cell differentiation. J. Biol. Chem. 2020, 295, 17413–17424.
- Sock, E.; Wegner, M. Using the lineage determinants Olig2 and Sox10 to explore transcriptional regulation of oligodendrocyte development. Dev. Neurobiol. 2021, 81, 892–901.
- Kageyama, R.; Shimojo, H.; Ohtsuka, T. Dynamic control of neural stem cells by bHLH factors. Neurosci. Res. 2019, 138, 12–18.
- Cai, J.; Chen, Y.; Cai, W.H.; Hurlock, E.C.; Wu, H.; Kernie, S.G.; Parada, L.F.; Lu, Q.R. A crucial role for Olig2 in white matter astrocyte development. Development 2007, 134, 1887–1899.
- Ju, J.; Liu, Q.; Zhang, Y.; Liu, Y.; Jiang, M.; Zhang, L.; He, X.; Peng, C.; Zheng, T.; Lu, Q.R.; et al. Olig2 regulates Purkinje cell generation in the early developing mouse cerebellum. Sci. Rep. 2016, 6, 30711.
- Li, H.; Richardson, W.D. Evolution of the CNS myelin gene regulatory program. Brain Res. 2016, 1641, 111–121.
- Yue, T.; Xian, K.; Hurlock, E.; Xin, M.; Kernie, S.G.; Parada, L.F.; Lu, Q.R. A critical role for dorsal progenitors in cortical myelination. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 1275–1280.
- Zhu, X.; Zuo, H.; Maher, B.J.; Serwanski, D.R.; LoTurco, J.J.; Lu, Q.R.; Nishiyama, A. Olig2-dependent developmental fate switch of NG2 cells. Development 2012, 139, 2299–2307.
- Lu, Q.R.; Sun, T.; Zhu, Z.; Ma, N.; Garcia, M.; Stiles, C.D.; Rowitch, D.H. Common Developmental Requirement for Olig Function Indicates a Motor Neuron/Oligodendrocyte Connection. Cell 2002, 109, 75–86.
- Takebayashi, H.; Nabeshima, Y.; Yoshida, S.; Chisaka, O.; Ikenaka, K.; Nabeshima, Y. The Basic Helix-Loop-Helix Factor Olig2 Is Essential for the Development of Motoneuron and Oligodendrocyte Lineages. Curr. Biol. 2002, 12, 1157–1163.
- Zhou, Q.; Anderson, D.J. The bHLH Transcription Factors OLIG2 and OLIG1 Couple Neuronal and Glial Subtype Specification. Cell 2002, 109, 61–73.
- Ligon, K.L.; Kesari, S.; Kitada, M.; Sun, T.; Arnett, H.A.; Alberta, J.A.; Anderson, D.J.; Stiles, C.D.; Rowitch, D.H. Development of NG2 neural progenitor cells requires Olig gene function. Proc. Natl. Acad. Sci. USA 2006, 103, 7853–7858.
- Mei, F.; Wang, H.; Liu, S.; Niu, J.; Wang, L.; He, Y.; Etxeberria, A.; Chan, J.R.; Xiao, L. Stage-specific deletion of Olig2 conveys opposing functions on differentiation and maturation of oligodendrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 8454–8462.
- Li, C.; Xie, Z.; Xing, Z.; Zhu, H.; Zhou, W.; Xie, S.; Zhang, Z.; Li, M.H. The Notch Signaling Pathway Regulates Differentiation of NG2 Cells into Oligodendrocytes in Demyelinating Diseases. Cell. Mol. Neurobiol. 2021.
- Tapson, V.F.; Torres, F.; Kermeen, F.; Keogh, A.M.; Allen, R.P.; Frantz, R.P.; Badesch, D.B.; Frost, A.E.; Shapiro, S.M.; Laliberte, K.; et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): A randomized controlled trial. Chest 2012, 142, 1383–1390.
- Munoz-Esquivel, J.; Gottle, P.; Aguirre-Cruz, L.; Flores-Rivera, J.; Corona, T.; Reyes-Teran, G.; Kury, P.; Torres, K.J. Sildenafil Inhibits Myelin Expression and Myelination of Oligodendroglial Precursor Cells. ASN Neuro 2019, 11, 1–10.
- Follis, R.M.; Carter, B.D. Myelin Avoids the JAM. Neuron 2016, 91, 713–716.
- Diez-Revuelta, N.; Higuero, A.M.; Velasco, S.; Penas-de-la-Iglesia, M.; Gabius, H.J.; Abad-Rodriguez, J. Neurons define non-myelinated axon segments by the regulation of galectin-4-containing axon membrane domains. Sci. Rep. 2017, 7, 12246.
- Schottlaender, L.V.; Abeti, R.; Jaunmuktane, Z.; Macmillan, C.; Chelban, V.; O’Callaghan, B.; McKinley, J.; Maroofian, R.; Efthymiou, S.; Athanasiou-Fragkouli, A.; et al. Bi-allelic JAM2 Variants Lead to Early-Onset Recessive Primary Familial Brain Calcification. Am. J. Hum. Genet. 2020, 106, 412–421.
- Marinho, W.; de Oliveira, J.R.M. JAM2: A New Culprit at the Pathophysiology of Primary Familial Brain Calcification. J. Mol. Neurosci. MN 2021, 71, 1723–1724.
- Jaggi, M.; Du, C.; Zhang, W.; Balaji, K.C. Protein kinase D1: A protein of emerging translational interest. Front. Biosci. 2007, 12, 3757–3767.
- Youssef, I.; Ricort, J.M. Deciphering the Role of Protein Kinase D1 (PKD1) in Cellular Proliferation. Mol. Cancer Res. MCR 2019, 17, 1961–1974.
- Omer, S.; Jin, S.C.; Koumangoye, R.; Robert, S.M.; Duran, D.; Nelson-Williams, C.; Huttner, A.; DiLuna, M.; Kahle, K.T.; Delpire, E. Protein kinase D1 variant associated with human epilepsy and peripheral nerve hypermyelination. Clin. Genet. 2021, 100, 176–186.
- Cen, C.; Luo, L.D.; Li, W.Q.; Li, G.; Tian, N.X.; Zheng, G.; Yin, D.M.; Zou, Y.; Wang, Y. PKD1 Promotes Functional Synapse Formation Coordinated with N-Cadherin in Hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 183–199.
- Fleming, C.E.; Saraiva, M.J.; Sousa, M.M. Transthyretin enhances nerve regeneration. J. Neurochem. 2007, 103, 831–839.
- Finsterer, J.; Wanschitz, J.; Quasthoff, S.; Iglseder, S.; Loscher, W.; Grisold, W. Causally treatable, hereditary neuropathies in Fabry’s disease, transthyretin-related familial amyloidosis, and Pompe’s disease. Acta Neurol. Scand. 2017, 136, 558–569.
- Alshehri, B.; Pagnin, M.; Lee, J.Y.; Petratos, S.; Richardson, S.J. The Role of Transthyretin in Oligodendrocyte Development. Sci. Rep. 2020, 10, 4189.
- Pagnin, M.; Dekiwadia, C.; Petratos, S.; Richardson, S.J. Enhanced re-myelination in transthyretin null mice following cuprizone mediated demyelination. Neurosci. Lett. 2022, 766, 136287.
- Ding, L.; Zhu, Z.; Wang, Y.; Zeng, L.; Wang, T.; Luo, J.; Zou, T.B.; Li, R.; Sun, X.; Zhou, G.; et al. LINGO-1 shRNA Loaded by Pluronic F-127 Promotes Functional Recovery After Ventral Root Avulsion. Tissue Engineerin. Part A 2019, 25, 1381–1395.
- Youssef, A.E.H.; Dief, A.E.; El Azhary, N.M.; Abdelmonsif, D.A.; El-Fetiany, O.S. LINGO-1 siRNA nanoparticles promote central remyelination in ethidium bromide-induced demyelination in rats. J. Physiol. Biochem. 2019, 75, 89–99.
- Wang, J.; Ye, Z.; Zheng, S.; Chen, L.; Wan, Y.; Deng, Y.; Yang, R. Lingo-1 shRNA and Notch signaling inhibitor DAPT promote differentiation of neural stem/progenitor cells into neurons. Brain Res. 2016, 1634, 34–44.
- Mi, S.; Hu, B.; Hahm, K.; Luo, Y.; Kam Hui, E.S.; Yuan, Q.; Wong, W.M.; Wang, L.; Su, H.; Chu, T.H.; et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat. Med. 2007, 13, 1228–1233.
- Sun, J.J.; Ren, Q.G.; Xu, L.; Zhang, Z.J. LINGO-1 antibody ameliorates myelin impairment and spatial memory deficits in experimental autoimmune encephalomyelitis mice. Sci. Rep. 2015, 5, 14235.
- Shao, Z.; Lee, X.; Huang, G.; Sheng, G.; Henderson, C.E.; Louvard, D.; Sohn, J.; Pepinsky, B.; Mi, S. LINGO-1 Regulates Oligodendrocyte Differentiation through the Cytoplasmic Gelsolin Signaling Pathway. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 3127–3137.
- Hanf, K.J.M.; Arndt, J.W.; Liu, Y.; Gong, B.J.; Rushe, M.; Sopko, R.; Massol, R.; Smith, B.; Gao, Y.; Dalkilic-Liddle, I.; et al. Functional activity of anti-LINGO-1 antibody opicinumab requires target engagement at a secondary binding site. mAbs 2020, 12, 1713648.
- Yang, C.; Tang, J.; Liang, X.; Qi, Y.; Luo, Y.; Xie, Y.; Wang, J.; Jiang, L.; Zhou, C.; Huang, C.; et al. Anti-LINGO-1 antibody treatment improves chronic stress-induced spatial memory impairments and oligodendrocyte loss in the hippocampus. Behav. Brain Res. 2020, 393, 112765.
- Ji, B.; Li, M.; Wu, W.T.; Yick, L.W.; Lee, X.; Shao, Z.; Wang, J.; So, K.F.; McCoy, J.M.; Pepinsky, R.B.; et al. LINGO-1 antagonist promotes functional recovery and axonal sprouting after spinal cord injury. Mol. Cell. Neurosci. 2006, 33, 311–320.
- Jepson, S.; Vought, B.; Gross, C.H.; Gan, L.; Austen, D.; Frantz, J.D.; Zwahlen, J.; Lowe, D.; Markland, W.; Krauss, R. LINGO-1, a transmembrane signaling protein, inhibits oligodendrocyte differentiation and myelination through intercellular self-interactions. J. Biol. Chem. 2012, 287, 22184–22195.
- Mi, S.; Pepinsky, R.B.; Cadavid, D. Blocking LINGO-1 as a therapy to promote CNS repair: From concept to the clinic. CNS Drugs 2013, 27, 493–503.
- Katanov, C.; Novak, N.; Vainshtein, A.; Golani, O.; Dupree, J.L.; Peles, E. N-Wasp Regulates Oligodendrocyte Myelination. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 6103–6111.
- Novak, N.; Bar, V.; Sabanay, H.; Frechter, S.; Jaegle, M.; Snapper, S.B.; Meijer, D.; Peles, E. N-WASP is required for membrane wrapping and myelination by Schwann cells. J. Cell Biol. 2011, 192, 243–250.
- Jin, F.; Dong, B.; Georgiou, J.; Jiang, Q.; Zhang, J.; Bharioke, A.; Qiu, F.; Lommel, S.; Feltri, M.L.; Wrabetz, L.; et al. N-WASp is required for Schwann cell cytoskeletal dynamics, normal myelin gene expression and peripheral nerve myelination. Development 2011, 138, 1329–1337.
- Bacon, C.; Lakics, V.; Machesky, L.; Rumsby, M. N-WASP regulates extension of filopodia and processes by oligodendrocyte progenitors, oligodendrocytes, and Schwann cells-implications for axon ensheathment at myelination. Glia 2007, 55, 844–858.
- Scaglione, A.; Patzig, J.; Liang, J.; Frawley, R.; Bok, J.; Mela, A.; Yattah, C.; Zhang, J.; Teo, S.X.; Zhou, T.; et al. PRMT5-mediated regulation of developmental myelination. Nat. Commun. 2018, 9, 2840.
- He, L.; Yu, K.; Lu, F.; Wang, J.; Wu, L.N.; Zhao, C.; Li, Q.; Zhou, X.; Liu, H.; Mu, D.; et al. Transcriptional Regulator ZEB2 Is Essential for Bergmann Glia Development. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 1575–1587.
- Quintes, S.; Brinkmann, B.G. Transcriptional inhibition in Schwann cell development and nerve regeneration. Neural Regen. Res. 2017, 12, 1241–1246.
- Wu, L.M.; Wang, J.; Conidi, A.; Zhao, C.; Wang, H.; Ford, Z.; Zhang, L.; Zweier, C.; Ayee, B.G.; Maurel, P.; et al. Zeb2 recruits HDAC-NuRD to inhibit Notch and controls Schwann cell differentiation and remyelination. Nat. Neurosci. 2016, 19, 1060–1072.
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Zeb2: A multifunctional regulator of nervous system development. Prog. Neurobiol. 2015, 132, 81–95.
- Weng, Q.; Chen, Y.; Wang, H.; Xu, X.; Yang, B.; He, Q.; Shou, W.; Chen, Y.; Higashi, Y.; van den Berghe, V.; et al. Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012, 73, 713–728.
- Quintes, S.; Brinkmann, B.G.; Ebert, M.; Frob, F.; Kungl, T.; Arlt, F.A.; Tarabykin, V.; Huylebroeck, D.; Meijer, D.; Suter, U.; et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 2016, 19, 1050–1059.
- Falcao, A.M.; Meijer, M.; Scaglione, A.; Rinwa, P.; Agirre, E.; Liang, J.; Larsen, S.C.; Heskol, A.; Frawley, R.; Klingener, M.; et al. PAD2-Mediated Citrullination Contributes to Efficient Oligodendrocyte Differentiation and Myelination. Cell Rep. 2019, 27, 1090–1102.e1010.
- Dreyton, C.J.; Knuckley, B.; Jones, J.E.; Lewallen, D.M.; Thompson, P.R. Mechanistic studies of protein arginine deiminase 2: Evidence for a substrate-assisted mechanism. Biochemistry 2014, 53, 4426–4433.
- Liu, Y.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Moore, E.; Goel, R.; O’Neil, L.; Mistry, P.; Hoffmann, V.; Mondal, S.; et al. Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus. JCI Insight 2018, 3, e124729.
- Musse, A.A.; Li, Z.; Ackerley, C.A.; Bienzle, D.; Lei, H.; Poma, R.; Harauz, G.; Moscarello, M.A.; Mastronardi, F.G. Peptidylarginine deiminase 2 (PAD2) overexpression in transgenic mice leads to myelin loss in the central nervous system. Dis. Models Mech. 2008, 1, 229–240.
- Mastronardi, F.G.; Wood, D.D.; Mei, J.; Raijmakers, R.; Tseveleki, V.; Dosch, H.M.; Probert, L.; Casaccia-Bonnefil, P.; Moscarello, M.A. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: A role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 11387–11396.
- Beniac, D.R.; Wood, D.D.; Palaniyar, N.; Ottensmeyer, F.P.; Moscarello, M.A.; Harauz, G. Cryoelectron microscopy of protein-lipid complexes of human myelin basic protein charge isomers differing in degree of citrullination. J. Struct. Biol. 2000, 129, 80–95.
- Yang, F.; Guan, Y.; Feng, X.; Rolfs, A.; Schluter, H.; Luo, J. Proteomics of the corpus callosum to identify novel factors involved in hypomyelinated Niemann-Pick Type C disease mice. Mol. Brain 2019, 12, 17.
- Yang, F.; Feng, X.; Rolfs, A.; Luo, J. Lovastatin promotes myelin formation in NPC1 mutant oligodendrocytes. J. Neurol. Sci. 2018, 386, 56–63.
- Yu, T.; Lieberman, A.P. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013, 9, e1003462.
This entry is offline, you can click here to edit this entry!