Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
The actin containing tropomyosin and troponin decorated thin filaments form one of the crucial components of the contractile apparatus in muscles. The thin filaments are organized into densely packed lattices interdigitated with myosin-based thick filaments. The crossbridge interactions between these myofilaments drive muscle contraction, and the degree of myofilament overlap is a key factor of contractile force determination.
- actin
- thin filament
- sarcomere
- length regulation
1. Introduction
Muscle contraction relies on the precise arrangement of myofibrils, composed of serially connected contractile units called sarcomeres. Sarcomeres are filamentous structures comprised of actin-based thin filaments and myosin-based thick filaments that slide along each other to generate force, ultimately leading to muscle contraction [1]. The force output of a muscle can be quantified as a function of myofilament overlap and is often characterized by the sarcomere length–tension relationship [2,3]. The length of the thick filaments is constant (~1.65 μm) and is conserved across vertebrate species and muscle types. However, the length of the thin filaments is variable, implying that thin filament length determines the extent of the overlap between the myofilaments, which is one of the important determinants of the amount of force generated. Accordingly, thin filament length correlates well with the physiological requirements of the muscle as slow-twitch fibers have longer thin filaments, while fast-twitch fibers have shorter thin filaments in general [4,5,6,7]. This observation also suggests that slow-twitch muscles have longer optimal sarcomere resting lengths, which might be one reason as to why they are able to efficiently maintain longer-term contractions. Therefore, to achieve optimal contraction characteristics, the thin filament length is expected to be precisely specified.
2. Thin Filament Structure
All muscles contain thin filaments (diameter of 6–10 nm) that consist of actin, tropomyosin (Tm), and troponin (Tn) in a 7:1:1 stoichiometry (Figure 1). Upon neuronal activation, Ca2+ is released from the sarcoplasmic reticulum, and triggers a conformational change in Tn, which shifts the azimuthal position of Tm on F-actin to allow for actin–myosin interactions. Over the past years, we have gained a good structural understanding of the thin filaments [13,14,15,16] that were analyzed by various advanced methods including X-ray fiber diffraction, electron microscopy (EM), and more recently, by cryo-EM [17,18,19,20,21,22,23,24,25]. Hence, the current models provide a highly resolved structure to provide insights into many aspects of their function and regulation (Figure 1). The structural core of the thin filament is the helical F-actin polymer that repeats once every 14 monomers and has an average axial repeat size of 36.5 nm (Figure 1). This structure is evolutionary highly conserved, although slight variations have also been reported. For instance, in the insect flight muscle, the average axial thin filament repeat size is 38.7 nm [26,27,28]. As much as the actin filaments themselves, the thin filaments are polarized structures with their barbed ends crosslinked in the Z-disc and capped by CapZ, and have their pointed ends extending toward the M-line where they are capped by tropomodulin (Tmod) (Figure 1). The thin filaments are stabilized by Tm molecules [29]; notably there are two pairs of Tm in every axial repeat, one on each side of the actin filament. The Tm dimers are linked end-to-end to form continuous α-helical coiled-coils that follow the F-actin long-pitch helix, where they regulate muscle contraction by sterically blocking the myosin target sites on the thin filament (Figure 1) [30]. Each Tm dimer has a regulatory heterotrimeric troponin complex attached to it, composed of troponin T (TnT), troponin C (TnC), and troponin I (TnI). TnT is the Tm binding subunit; TnC is the Ca2+-binding regulatory subunit; and TnI is the inhibitory subunit (Figure 1) (for a recent review see: [31]).
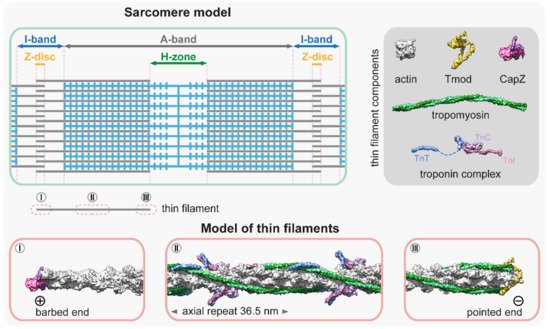
Figure 1. Molecular representation of the thin filaments. The schematic at the top left is a simplified sarcomere model with thick (blue) and thin filaments (gray), and the major sarcomeric regions. The dark gray box at the top right shows the main components of the thin filaments [13,14,32,33,34,35]—PDB: 5JLF, 4PKI, 4PKG, 7PDZ, 1C1G, 4Y99, 1J1E, 2Z5H. The molecular models below depict three representative thin filament regions: (I) The thin filament proximal to the barbed end is not decorated with tropomyosin and troponin, however, it is associated with other factors (elastic filaments, nebulin, Z-disc proteins, etc.) that are not depicted in this representation. The barbed end is capped with a CapZ heterodimer [34]; PDB: 7PDZ. (II) The central region is associated with tropomyosin–troponin complexes [25]; PDB: 7KO4. This section depicts a full axial repeat. Note that in some muscles, this part of the thin filament also contains two nebulin molecules. (III) The pointed end is capped by tropomodulin (Tmod) [33]; PDB: 4PKI, 4PKG. The association of Tmod to the pointed end is significantly enhanced by tropomyosin binding.
Conventional structural reconstructions provide an averaged image of the thin filaments, however, it has long been known that thin filaments are not perfectly uniform [36,37]. It has been shown that the terminal units of the actin filaments adopt a conformation that is different to the rest of the filament [38,39]. Moreover, actin-binding proteins can exert conformational changes on the filaments (i.e., formin binding to the barbed end can induce a more flexible conformation through long-range allosteric effects [40,41]; and ADF/cofilins binding near the pointed end can shorten the helical pitch of the filaments [42,43,44]). Therefore, the structure of the ends of the thin filaments is likely to differ from the averaged reconstructions, and it might provide us with essential cues to better understand the mechanisms of filament length regulation.
In situ cryo-electron tomography and cryo-focused ion-beam scanning electron microscopy analysis allow for the molecular and structural analysis of thin filaments in their native environment and have been proven to be powerful enough to explore the local structural differences at the single sarcomere and even at the single thin filament levels [45,46]. For example, it was revealed that the position of Tm differs in the I-band from that in the A-band, where the binding of myosin to the thin filament shifts Tm from the C-state to the M-state. Tm in the I-band remains in the C-state and the transition happens in one Tm unit immediately after the A-band/I-band transition [46]. Nevertheless, the use of these techniques has remained limited in regions of the Z-disc and the M-band/H-zone, where a large number of regulatory and structural proteins are present, forming pleomorphic densities on the EM images, thereby prohibiting the identification of individual proteins and precise reconstruction of the filament ends. One alternative to resolve the composition and arrangement of these dense sarcomeric regions can be fluorescent nanoscopy, as was recently demonstrated [47,48]. Single-molecule localization microscopy combined with structure averaging allows for the determination of the position of sarcomeric proteins with a precision of <10 nm, and it has been successfully used to reconstruct various large protein complexes (for a recent review see [49,50]). Future studies using the combination of these new methods will hopefully provide us with superior molecular models of the pointed and barbed ends of the thin filaments, which is essential to a better understanding of their dynamics and length regulation.
3. Thin Filament Assembly
The backbone of the thin filament is an F-actin molecule, which is assembled from a G-actin monomer pool. Actin monomers polymerize spontaneously under physiological salt conditions starting with a slow nucleation step, in which a few actin monomers combine to form a nucleus or ‘seed’ for subsequent elongation (Figure 2). The association of the monomers is fast, but the dimers and trimers are very unstable. The nucleation or ‘seed’ formation step is followed by elongation in which monomers bind to (and dissociate from) the two ends of the filaments. Elongation has different characteristics on the opposite ends of the filament as barbed ends grow much faster than pointed ends with a significantly lower “critical concentration” (~0.1 µM and ~0.7 µM, respectively), and elongation is favored when the concentration of free G-actin exceeds the critical concentration (Figure 2) [51,52].
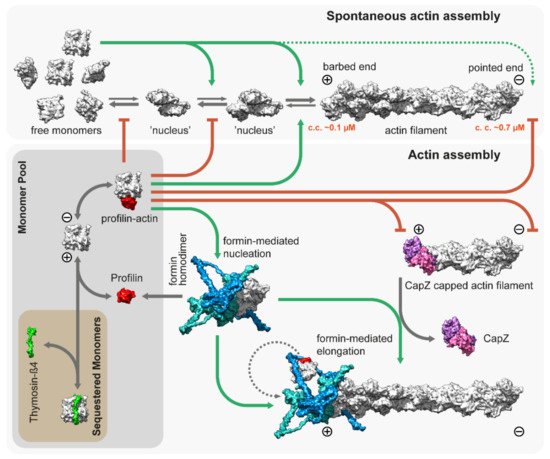
Figure 2. Spontaneous and regulated assembly of actin filaments. Free G-actin monomers can form nucleation seeds, which can elongate rapidly from both their barbed and pointed ends, although the critical concentration is significantly lower for the barbed ends (~0.1 µM). Thymosin-β4 sequesters actin monomers and prohibits uncontrolled actin assembly in vivo. Similarly, profilin [53]; PDB: 2BTF, bound to actin monomers, inhibits both spontaneous nucleation and growth from the pointed end. However, it allows for barbed end association and feeds actin monomers to formins, which are actin nucleation and elongation factors. The figure depicts the template-based [54,55] structure of DAAM1 using multiple PDB models [56,57,58]; PDB: 2Z6E, 2V8F, 1Y64. CapZ bound to the filaments prevents growth from the barbed end [34]; PDB: 7PDZ.
Actin is an abundant protein with a concentration well above the critical concentration in non-muscle cells, ranging from 50 to 200 µM [59,60]. In embryonic muscle cells, almost half of the actin pool is in a monomeric form, but the G-actin concentration decreases significantly during development; in adult muscles, it is just above the critical concentration (~0.2–0.3 µM) for barbed ends [61,62]. In addition, most of the actin monomers are bound either to profilin or thymosin-β4, which shields the barbed end side of actin monomers and prevents spontaneous nucleation [53,63,64]. While thymosin-β4 completely sequesters G-actin, thereby preventing both nucleation and elongation, profilin only inhibits nucleation and pointed end elongation. Profilin–actin complexes can bind to the exposed filament barbed ends and following its incorporation, profilin dissociates from the actin protomer (Figure 2). Muscle tissues express both profilin and thymosin-β4 [65,66], however, their role in thin filament assembly and elongation has mostly been unexplored. Nevertheless, one study demonstrated that overexpression of profilin resulted in thin filament and sarcomere elongation in the flight muscles of Drosophila [67], suggesting a promoting role in thin filament formation. Since nucleation is the rate limiting step of actin assembly, cells use nucleation factors to form and stabilize polymerization nuclei. While the branched actin filaments are nucleated by the Arp2/3 complex, the unbranched filaments such as the thin filaments are initiated by formins. It is possible that the WASP-homology 2 (WH2) domain containing “nucleators” (such as Spire, Cordon-bleu, or Leiomodin) also play a role in thin filament assembly, although it is unlikely that they could mediate this process without formins [68]. Formins are highly conserved, multidomain proteins characterized by the presence of two formin homology domains (FH1 and FH2) [69]. The FH2 domains can directly bind G- and F-actin and are able to nucleate actin filaments by stabilizing actin dimers [70], thus overcoming the kinetic barrier of nucleation (Figure 2). After nucleation, formins can remain bound to the barbed end of the filaments and promote their processive elongation [71,72]. This “processive capping” allows formins to protect the growing barbed end from the inhibitory effects of capping proteins such as CapZ (Figure 2) [73,74,75]. The flexible FH1 domains can interact with profilin–actin complexes and provide new subunits to the growing filament [76,77,78,79]. Additionally, formins possess other activities including the ability to bind along the lengths of actin filaments or microtubules, promoting bundling and crosslinking or coordination [80,81,82,83,84,85,86,87].
Eukaryotic species have multiple formin proteins and it has been shown that in vertebrates, 13 out of the 15 formin genes are expressed during postnatal heart development [88], while in the Drosophila flight muscle, all of the six fly formins are expressed at some point of development [89]. Among the sarcomeric formins, members of the FHOD family have been the most extensively studied. While FHOD1 is expressed in both the muscle and non-muscle cells, in cardiomyocytes, FHOD1 primarily localizes to the costameres and the intercalated discs, but it is largely excluded from the sarcomeres [90,91]. In contrast, FHOD3 is abundantly expressed in the heart muscle and it displays a mainly sarcomeric accumulation [92]. Silencing of FHOD3 leads to the disruption of myofibrils in cultured cardiomyocytes [93,94], and consistently, FHOD3 deficient mice die prenatally due to failed myocardial development [95]. Detailed analysis revealed that these mutant cardiomyocytes are able to form premyofibrils, however, they fail to mature into myofibrils [95]. In humans, FHOD3 mutations have been associated with hypertrophic and dilated cardiomyopathies [96,97,98,99]. In addition, FHOD3 has been shown to play a maintenance role in the adult mouse heart [100]. Similarly, muscle specific silencing of Fhos, the single Drosophila FHOD orthologue, severely disrupts the organization of the flight muscle myofibrils, while allowing for the proper initiation of muscle fiber development [101]. Silencing of Fhos in a later phase of flight muscle myofibrillogenesis resulted in sarcomeres with significantly narrower widths, suggesting that Fhos is necessary for the peripheral (radial) growth of the thin filaments. Despite these advances, the molecular mechanisms of FHOD type formins remain largely obscured. In bulk polymerization assays using sarcomeric actin, both FHOD1 and FHOD3 were unable to nucleate actin filaments [93,102], although it was later demonstrated that FHOD1 can nucleate actin filaments from cytoplasmic actin [103]. Furthermore, the findings on the localization of FHOD3 in myofibrils is also somewhat controversial. Most studies have shown that FHOD3 is localized to broad stripes in the A-band, more precisely, in the C-zone [93,94,95,100,104,105], and it was demonstrated that this localization is dependent on a direct interaction with the thick filament associated MyBP-C protein [105]. However, a FHOD3 enrichment has also been detected at the Z-discs in the mouse and human adult heart sections and in the extracted myofibrils [88,94]. In contrast to FHOD, Drosophila Fhos is able to nucleate both cytoplasmic and sarcomeric actin, it allows elongation in the presence of profilin, and it protects the barbed ends from capping protein binding [103]. Accordingly, Fhos is localized to the Z-disc [101] into the immediate vicinity of the barbed ends, as suggested by nanoscopic analysis [47]. Surprisingly, however, flies with a mutation abolishing the barbed end binding and nucleation activity of Fhos have small (short and thin) but properly organized sarcomeres [101]. Based on these observations, the common feature of FHOD type formins is that they are not required for the synthesis of the initial pool of sarcomeric thin filaments. Furthermore, their sarcomeric localization and biochemical activities suggest that their barbed end binding activities are only secondary or negligible, and therefore FHOD type formins may primarily use their actin side binding and bundling activity to organize pre-existing filaments into ordered thin filament arrays during myofibrillogenesis. Nevertheless, FHOD dependent actin assembly is required to achieve the mature sarcomere size.
4. Thin Filament Elongation
After their initial assembly, thin filaments must elongate to achieve their well-defined length, fitting best to the workload of a given muscle type [115]. In addition, they need to be able to elongate in response to mechanical strain to achieve the optimal efficiency, as observed in mature cardiac muscles [116]. Fluorescently labeled actin monomer incorporation studies in chick cardiac myocytes revealed that in muscle cells, both ends of the thin filaments are dynamic, however, the subunit exchange at the pointed end is significantly higher [117]. Interestingly, the blocking of barbed end dynamics did not arrest actin filament elongation, while the blocking of pointed end dynamics was sufficient to prevent filament growth in multiple model systems [117,118]. Furthermore, it was also demonstrated that labeled Tm also incorporates at the pointed end in rat cardiac myocytes [119]. Therefore, it was concluded that, in contrast to non-muscle cells, actin filaments somehow elongate from their pointed end during myofibrillogenesis [117,118].
The thin filaments are thought to be capped at both ends; the barbed end is associated with the capping protein (CapZ) complex, and the associated regulatory factors maintain the dynamics of monomer exchange [120], while the pointed end is capped by Tmod (Figure 1). Tmods (Tmod1–Tmod4 in vertebrates) are conserved proteins that have a distinct role in length regulation of the thin filaments [121,122,123]. A single Tmod molecule interacts with the thin filament via two actin binding sites (ABS1 and ABS2) and two Tm binding sites. Tropomyosin binding enhances its pointed end capping activity by 5- to 10-fold, and once Tmod is fully bound, it blocks the addition of actin monomers to the pointed end and prevents filament elongation as well as depolymerization [33,124]. Consistently, the silencing of Tmod or impairing of its capping activity leads to thin filament lengthening in various muscle tissues [123,125,126,127]. Conversely, the overexpression of Tmod inhibits actin incorporation at the pointed end, leading to thin filament shortening. Hence, thin filament length has an inverse proportion to the expression level of Tmod, as shown in the sarcomeres of cardiac myocytes and Drosophila flight muscles [117,118]. However, Tmod binding to the pointed end is dynamic, and it was initially suggested that Tmod allows the non-catalyzed addition of G-actin to the pointed end by acting as a leaky cap, leading to filament elongation (for a review, see [128]. In this scenario, thin filament elongation would be regulated exclusively by the expression level of Tmod.
Independent of its precise mode of action, Tmod is considered as a negative regulator of actin elongation, whereas members of the highly homologous Leiomodin (Lmod) protein family were discovered as promoters of thin filament elongation [129,130]. The Lmod family consists of three isoforms in vertebrates (Lmod1–Lmod3), which are expressed primarily in the muscle cells and behave as strong actin nucleators in biochemical assays [129,131,132]. Lmod2 and 3 are both expressed in striated muscles with Lmod2 being the predominant isoform in cardiac muscles and Lmod3 the major isoform in skeletal muscles. Defective Lmod2 gene expression leads to dilated cardiomyopathy both in mice and humans [115,133,134], while the Lmod3 gene is associated with nemaline myopathy [131,135]. Loss of Lmod1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS) in both humans and mice [136]. Initially, Lmods were proposed to be the long-sought-after muscle actin nucleators [129], however, loss of function studies and the developmental timing of their expression revealed that Lmods are unlikely to be required during the initial phases of thin filament assembly [137]. Overexpression of Lmod2 leads to the elongation of thin filaments [130,138], and its loss results in thin filament shortening [115,139]. Hence, it was proposed that Lmod2 is most likely to be involved in thin filament elongation and/or maintenance. While Lmods are essential factors in vertebrate muscles, they are not present in Drosophila. Curiously, a nonrelated protein, called sarcomere length short (SALS)-, displaying a number of functionally similar properties, has been found in Drosophila. Like Lmods in vertebrates, SALS promotes thin filament elongation and sarcomere growth in primary Drosophila muscle cell cultures and in the flight muscles of Drosophila [101,140]. Loss of SALS causes shortening of the thin filaments and lack of actin incorporation at their pointed ends. The effect caused by Tmod overexpression was also increased by the loss of SALS, which suggests that Tmod and SALS antagonize each other functionally at the filament pointed ends during thin filament elongation [101,140].
Despite the role of Lmods having been extensively studied over the last decade, no consensus has been reached on their exact molecular mechanisms; instead, two prominent but contradicting models have emerged to explain the in vivo and in vitro observations [128,141]. The ‘competition model’ suggests that thin filament elongation is regulated by the interplay between the pointed end capping Tmod and its functional antagonist Lmod, which can displace Tmod from the pointed end and promote catalyzed or non-catalyzed subunit addition (Figure 3). As an alternative, the ‘nucleation model’ suggests that Lmod nucleates new filaments, which can then anneal to the pointed ends of existing filaments or integrate into the sarcomeres during myofibril maturation and/or during repair (Figure 3). It has also been suggested that Lmod (and SALS) could cooperate with muscle-specific formins, which would mediate the processive elongation of these newly nucleated ‘mini filaments’ [109]. Part of the controversy surrounding the molecular functions of Lmods is from the fact that due to technical difficulties, the current structural and biochemical data are obtained by using Lmod fragments (instead of the full length protein) interacting with actin monomers and Tm. Results obtained from these experiments are difficult to interpret since truncated protein fragments are often insufficient to reconstitute the biological function of a full-length protein. In spite of this limitation, previous studies have exploited the modular organization of Lmods to assign functions to every domain and to decipher the function of the native protein. Lmods share several domains with Tmods, but they also have a C-terminal WH2 and a proline-rich domain (PRD) containing the extension. Lmods have only one Tm binding site, and NMR spectral analysis suggests that it folds into an α-helical hairpin, which is positioned over the N-terminus of Tm [138]. Since unobstructed Tm N-termini are only available at the pointed ends, it was interpreted as evidence supporting the “competition model”. However, a free Tm N-terminus should also be available during thin filament nucleation, therefore, it does not refute the “nucleation model”. In any case, Tm binding was shown to be important in vivo as the reduced binding affinity of this motif blocked the thin filament elongation effect of exogenous Lmod2 in cardiomyocytes [138]. ABS1 is essential for pointed end capping in Tmods and its sequence is also relatively well conserved in Lmods. However, it is controversial whether it has an actin binding activity in Lmods [132,138,142]. ABS2 is structurally similar in Tmods and Lmods, however, they appear to fulfill different functions. In Lmod, the ABS2 domain lacks the DNase I-binding loop, which is important for pointed end capping, and the ABS2 in Lmod can bind two or three actin subunits on its own, making it primarily responsible for the nucleation activity of Lmod [132]. Lmods also contain a PRD, which is an ideal candidate for profilin–actin binding, however previous studies have concluded that it is not able to bind profilin [129,132]. Similarly, the proline-rich domain of SALS is most likely not involved in profilin binding as profilin does not influence the activities of the actin binding domains of SALS [143]. Alternatively, the PRD might mediate an interaction with SH3 domains to regulate subcellular localization or activity, as has been suggested for other muscle proteins [144]. Finally, Lmods contain a C-terminal extension with a WH2 domain, which is able to bind G-actin in the barbed end groove [145], and in general, it is thought to function as an actin monomer recruiting motif in numerous other actin binding proteins [146]. At first, it was thought to play an essential role in nucleation [129,130], but later it turned out to be only secondary to ABS2 [132]. Moreover, the C-terminal extension has a more pronounced role in nucleation than the WH2 domain on its own [145]. In contrast, SALS contains a tandem WH2 domain, which is assumed to exclusively mediate its interaction with F-actin and G-actin [140,143]. Aside from the PRD and the WH2 domains, SALS is proposed to be mostly unstructured, which hinders its biochemical analysis.
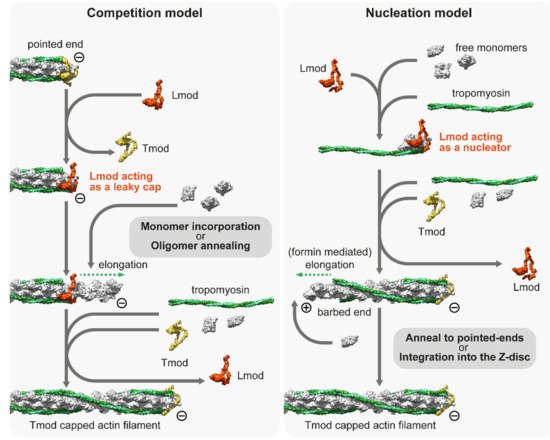
Figure 3. The competition and nucleation models of Leiomodin (Lmod). The competition model suggests that Lmod competes with Tmod, and once it is bound to the pointed end, it acts as a leaky cap, allowing the addition of monomers or short oligomers to the pointed end. The nucleation model suggests that Lmod is not able to bind to the pointed end but can act as a nucleation factor by binding at least three actin monomers and a tropomyosin dimer. The model proposes that Lmod dissociates from the actin nucleus when it begins to elongate, which can be mediated by formins. The newly formed filament can be incorporated into the Z-disc or annealed to free pointed ends. Note that both models presume that free monomers are available. The molecular model of Lmod is based on the tentative structure proposed by Tolkatchev et al. [138].
The other crucial question as to the mechanisms of Lmod is whether it localizes to the pointed ends in vivo, as it is a key part of the “competition model”. Originally, it was demonstrated that Lmod is localized to the H-zone of cultured rat cardiomyocytes [129], though the pointed ends were not resolved in that study. Following that, it was demonstrated that Lmod localizes at or close to the pointed end in isolated myocytes [130,131]. They also found that intensities of Tmod1 and Lmod2 often seemed to have an inverse correlation, suggesting mutually exclusive pointed end binding [130]. However, later studies showed that Lmod is not restricted to the pointed end as it is localized to a broader region within the A-band, suggesting that Lmod might bind the sides of the thin filaments [131,137,147,148,149]. Moreover, the studies demonstrating the Tmod–Lmod overlap used diffraction limited fluorescent microscopy, which was not able to provide sufficient resolution to settle this question. Similar to Lmods, SALS is mostly localized near the H-zone during flight muscle development in Drosophila [140] and a recent nanoscopic analysis revealed that there was less than a 2 nm difference in the average localization of Tmod and the WH2 domain containing central region of SALS [47]. This strongly suggests that SALS is localized to the pointed end where it probably interacts/competes with Tmod, which at least in the case of SALS supports the “competition model”. A similar analysis using an appropriate vertebrate model could ultimately put an end to the controversy and determine whether Lmods are pointed end binding proteins, thin filament side binding proteins, or localized to a more central part of the H-zone.
Aside from Lmods, N-WASP—another WH2 containing actin “nucleator”—has also been linked to myofibrillogenesis as a cooperating partner of nebulin [144]. It was suggested that in response to IGF1, N-WASP is targeted to the Z-disc by nebulin, where it promotes actin incorporation and thus filament formation as a hypertrophy response [144]. However, later studies were not able to reproduce this result [150,151]. Furthermore, the suggested barbed end actin incorporation would contradict the more established pointed end elongation model of thin filaments.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23105306
This entry is offline, you can click here to edit this entry!