Pancreatic cancer has the worst prognosis and lowest survival rate among all types of cancers and thus, there exists a strong need for novel therapeutic strategies. Chimeric antigen receptor (CAR)-modified T cells present a new potential option after successful FDA-approval in hematologic malignancies, however, current CAR T cell clinical trials in pancreatic cancer failed to improve survival and were unable to demonstrate any significant response. The physical and environmental barriers created by the distinct tumor microenvironment (TME) as a result of the desmoplastic reaction in pancreatic cancer present major hurdles for CAR T cells as a viable therapeutic option in this tumor entity. Cancer cells and cancer-associated fibroblasts express extracellular matrix molecules, enzymes, and growth factors, which can attenuate CAR T cell infiltration and efficacy. Recent efforts demonstrate a niche shift where targeting the TME along CAR T cell therapy is believed or hoped to provide a substantial clinical added value to improve overall survival.
- tumor microenvironment
- pancreatic cancer
- immunotherapy
- CAR T cell therapy
- extracellular matrix
- cancer-associated fibroblasts
- Introduction
Pancreatic cancer, i.e., pancreatic ductal adenocarcinoma (PDAC), is a fatal disease with five-year overall survival rates of 1% to 5% and median survival duration of fewer than six months [1]. The poor prognosis has not substantially changed during the past decades, establishing pancreatic cancer as the fourth leading cause of cancer-related deaths in Western countries [2–4]. Therapeutic progress in other types of cancer will lead to its ascension in second place among all cancers within the next decade [5]. Surgery remains the only potentially curative treatment, but only a minority of patients show a resectable disease stage at diagnosis, due to invasion to the surrounding vasculature and due to lack of symptoms at an early stage [6]. Nonetheless, the median overall survival is still only 24 months for patients with resectable disease [7].
Therapeutic failures of chemotherapy, targeted therapy, and immunotherapy of PDAC can be largely attributed to the special features of this cancer, which exhibits highly nutrient-poor, immunosuppressive, hypoxic and desmoplastic characteristics leading to rapid cancer progression [8]. The tumor is composed of only a minor number of malignant cells within a microenvironment of dense extracellular matrix (ECM), a barrier that prevents adequate drug delivery and might serve as a prognostic factor ( Figure 1; Figure 2) [8]. Responsible for the stromal reaction are mainly cancer-associated fibroblasts (CAFs) that develop from bone marrow-derived mesenchymal stem cells (MSCs), pancreatic stellate cells (PSCs), and quiescent resident fibroblasts through multiple pathways of activation [9]. The complex tumor vasculature in PDAC is characterized by a lack of blood vessels, leading to high levels of hypoxia in the tumor interior [10]. Furthermore, the capillaries and lymphatic vessels that are present tend to be collapsed due to high interstitial pressure, either from excess fluid or from solid stress [11]. Other non-neoplastic cancer-associated cells consist of immune-suppressor cells such as regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), and tumor-associated macrophages (TAM) that can inhibit CD8+ T cells, which play a key role in the antitumor immune response, and thereby establish an immunosuppressive tumor microenvironment [12]. Neural remodeling and perineural invasion (PNI), the neoplastic invasion of tumor cells into nerves, are further unfavourable histological features, and are considered as one of the main routes for cancer recurrence and metastasis after surgery [13]. Conventional therapies such as chemotherapy and radiation have focused on effective therapy of the malignant cell population. Thus, a concordant combination of various treatments targeting additional key cellular features of PDAC such as stroma, reversing suppressive immune reactions and enhancing antitumor reactivity may lead to more successful treatment strategies [14]. Thus, there is a clinically unmet need for new therapeutic options.
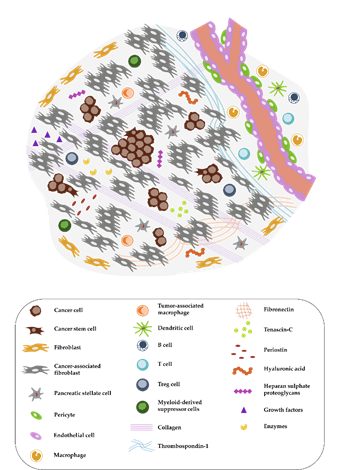
Figure 1. Complex tumor microenvironment (TME) of pancreatic cancer. The pancreatic ductal adenocarcinoma (PDAC) microenvironment is characterized by a dense desmoplastic stroma, with cancer-associated fibroblasts (CAFs) presenting the majority of the cell population (in grey). Tumor cells (round and brown) in aggressive PDACs can occur in tumor buds, small groups of cells, especially in the invasive front. A high abundance of extracellular matrix (ECM) molecules, enzymes, and growth factors is another important feature. Immune cells are often excluded from the TME or exhibit an immunosuppressive phenotype. The distribution of pro- and anti-inflammatory immune cells as well as the exact composition of the tumor stroma is dependent on the subtype of pancreatic cancer as discussed by Bailey et al. or by Karamitopoulou [12,15].
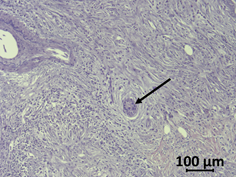
Figure 2. Haematoxylin/eosin-stained human PDAC sample. Tumor cells (arrow) are surrounded by the desmoplastic reaction of stromal cells and few immune cells.
- CAR-T cells
Immunotherapy is a rapidly developing field within oncological research, especially since the development of chimeric antigen receptor (CAR) T cells, which are genetically engineered to express receptors targeting cancer cells for immunotherapy. CAR technology has made leaps of development since its conception in 1993, combining antigen recognizing regions from antibodies with intracellular T cell signaling domains (Figure 3) [16]. In this way, potential demasking of tumor cells by major histocompatibility complex (MHC) class I downregulation, can be overcome [17]. At first, double chimeric receptors were developed by engineering the VH and VL chains of immunoglobulins to the constant regions of the T cell receptor (TCR) [18]. Over time, CARs were modified into a single chain approach coupling a single chain variable fragment (scFv) derived from an antibody via a spacer and transmembrane domain to the CD3ζ signaling domain of the TCR [16]. The addition of costimulatory domains from CD28 or 4-1BB generated a stronger activating signal, circumventing the intracellular activation by TCR-domains, defining the second CAR generation [19]. Second-generation CARs targeting CD19 are the first CAR success-story wherein phase II study 81% of the B cell acute lymphoblastic leukemia patients demonstrated complete remission 28 days after infusion [20]. Their tremendous success in the treatment of leukemia and lymphoma patients led to the FDA approval of the first CAR T cell therapy as a second-line treatment in 2017 [21]. The incorporation of further costimulatory domains derived from CD27 or CD40 as well as the introduction of additional cytokine expression or induction of other signaling pathways established the third, fourth, and fifth generations of CAR T cells, increasing cytokine production, cell survival, and persistence [22]. In recent years, advanced CAR concepts, such as Tandem or Universal CAR approaches have been developed and enabled the targeting of challenging antigen expression profiles on cancer cells [23]. Other advanced CAR technologies explore mechanisms to switch on and off CAR expression on T cells to control possible toxic side effects [24]. Another upcoming class of engineered receptors is synthetic Notch (synNotch) receptors, which can induce transcriptional activation after target recognition [25]. Ultimately, all developmental generations of CARs offer various opportunities and challenges for prospective cell-based approaches as reviewed before [22,24,26].
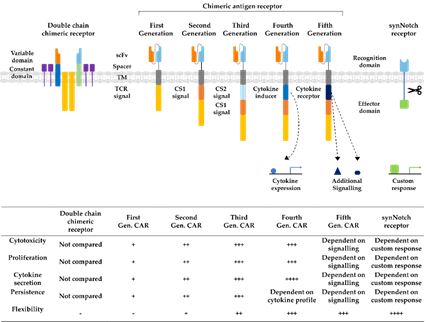
Figure 3. Developmental stages of chimeric antigen receptors. The first double chain chimeric receptors were engineered to customize the variable T cell receptor (TCR) domain by using VH and VL chains of antibodies (orange and bright blue boxes) fused to the constant regions of the TCR α- and β-chains (green and blue boxes). They mimicked the TCR in appearance and functionality. Activation relies on association with intracellular CD3ζ (yellow boxes), CD3γ, CD3δ, and CD3ε chains (purple boxes). The first generation of CARs combined the antigen recognizing scFv directly with the CD3ζ-signalling domain in one construct overcoming expression difficulties by the tremendous construct length of double chain chimeric receptors. Cytotoxicity, proliferation, cytokine secretion, and persistence of CARs were increased in second and third generation CARs by the addition of further costimulatory domains (CS1 and CS2) such as CD27, CD28, CD134, or 4-1BB. Introduction of T cell redirected for universal cytokine-mediated killing (TRUCKs) or fourth generation CARs increased the flexibility in CAR design for specific challenges even further, enabling local expression of cytokines such as IL-12, which are toxic in high concentrations. Fifth generation CARs, as fourth generation CARs, are based on second generation CARs. The individual antigen response is complemented by activation of intracellular domains of cytokines (dark blue box) e.g., IL-2Rβ, which induced signal transduction in the STAT3/5 pathway. Another group of artificial antigen receptors, gaining increased interest in recent years, are synNotch receptors. These receptors use the cleavage process after Delta-Notch binding and enable an unlimited variety of responses (green box) after target recognition such as cell fate determination with transcription factors and expression of selected cytokines or therapeutic antibodies. In this way, they bring the potential of immune cells as “living drugs” a big step forward.
Unfortunately, fewer exciting outcomes were achieved in initial clinical trials with CAR T cells targeting solid tumors, including PDAC. Successful CAR therapy for carcinomas needs to overcome the physical and environmental barriers in the tumor microenvironment (TME) [27]. The TME consists, next to tumor cells, of endothelial, immune, and inflammatory cells, stromal cells, the extracellular matrix and a broad spectrum of enzymes, cytokines, and growth factors [28]. This creates a strong physical barrier for CD8+ T cells, while their immune response is further diminished by the high amount of immunosuppressive immune cells present in the TME of PDAC [12,29]. These aspects must be considered and addressed in the field of cell-based immunotherapy against solid cancers.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12061389