The success of conventional chimeric antigen receptor (CAR) therapy in the treatment of refractory hematologic malignancies has triggered the development of novel exciting experimental CAR technologies. Among them, adaptor CAR platforms have received much attention. They combine the flexibility and controllability of recombinant antibodies with the power of CARs. Due to their modular design, adaptor CAR systems propose answers to the central problems of conventional CAR therapy, such as safety and antigen escape.
- chimeric antigen receptor (CAR)
- adaptor molecule
- adoptive T cell therapy
- cancer immunotherapy
- Introduction
Cancer immunotherapy is a rapidly growing field that is becoming more and more important in clinical practice. The development of chimeric antigen receptor (CAR) T cells, formerly known as T-bodies [1], revolutionized adoptive cell transfer. To date, over 520 clinical trials have emerged worldwide, redirecting CAR T cells against 64 different tumor targets [2]. Among them, two CD19-specific CAR T cell products are approved for the treatment of acute lymphoblastic leukemia (ALL) and large B cell lymphoma [3–5]. Except for CD19 CARs, the most promising results have currently been achieved for the targeting of CD22 [6] and B cell maturation antigen (BCMA) [7,8] in ALL and multiple myeloma, respectively. However, driven by the selective pressure of mono-specific CAR T cells, antigen-negative escape variants frequently occur and often impede the initial success of these living drugs (e.g., [6,9–12]). The overall translation of CAR technology to non-hematologic malignancies remains challenging. Physical barriers and the immunosuppressive microenvironment of solid tumors represent major obstacles, as they impair CAR T cell migration and function [13]. Low levels of target antigen expression in healthy tissues can result in severe “on-target, off-tumor” toxicities, as exemplified by the occurrence of lethal pulmonary toxicity in a colon cancer patient after the infusion of autologous α-HER2/neu CAR T cells [14]. Besides the damage of healthy tissues, cytokine release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES) are frequently observed side effects of CAR T cell therapy [15].
In order to overcome the above-mentioned limitations, the scientific community is pursuing various new CAR concepts and models [13]. This review will focus on recent advances in the field of switchable adaptor CAR platforms, with emphasis on their safety profile, controllability, target flexibility, specificity, and efficiency.
- Adaptor CAR Design
CARs are artificial receptors composed of three key elements: an extracellular (tumor binding) domain followed by a transmembrane and intracellular signaling domain(s) (Figure 1a) [16,17]. Similar to antibodies, CAR-modified T cells are able to bind naturally occurring surface molecules independent of their own T cell receptor (TCR). Upon tumor recognition, downstream signaling pathways are activated, triggering the lysis of malignant cells [16,17]. Over the last decades, CAR design has undergone continuous changes from 1st to 4th generation that have improved CAR T cell expansion, cytotoxicity, cytokine secretion, and in vivo persistence [18].
Adaptor CAR T cells were developed with the aim to improve the flexibility, tumor specificity, and controllability of conventional CAR T cells. To achieve these objectives, the tumor-targeting and signaling moieties of conventional CARs were uncoupled, resulting in a dichotomous system consisting of an adaptor CAR and soluble, tumor-specific adaptor molecules (Figure 1b). The basic structure of adaptor CARs corresponds to the conventional CAR design (Figure 1a), although the extracellular domain does not interact with a tumor-associated antigen but with a binding partner incorporated into the adaptor molecule. The bifunctional adaptor molecule in turn provides tumor specificity and acts as a linker at the interface between the tumor and the adaptor CAR T cell. This complex can then mediate anti-tumor responses, similar to conventional CAR T cells (Figure 1c).
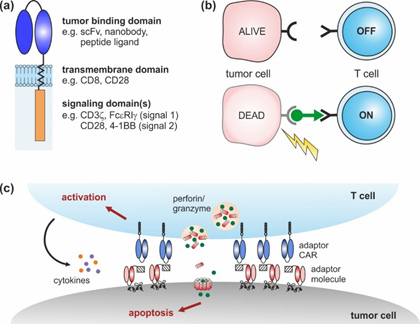
Figure 1. Chimeric antigen receptor (CAR) design: (a) conventional CARs are composed of an extracellular tumor binding domain followed by a transmembrane and intracellular signaling domains. (b) Adaptor CAR systems are composed of T cells engineered with an adaptor CAR and soluble adaptor molecules (green). Adaptor CAR T cells are per se inactive (OFF). In the presence of tumor-specific adaptor molecules, they are turned ON. (c) Upon cross-linkage via adaptor molecules, adaptor CAR T cells elicit potent anti-tumor responses that finally result in tumor lysis.
Notably, the dual principle of adaptor CAR systems provides an important molecular safety switch to precisely control the adaptor CAR T cell activity. Anti-tumor responses will decrease and vanish with the elimination of the adaptor molecule from the body. Vice versa, repeated adaptor molecule administration will permit the re-initiation of therapy against the same or an alternative target in case of tumor relapse. Overall, the adaptor CAR T cell activity as well as the associated side effects (e.g., CRS, “on-target, off-tumor” toxicity) might be controlled in a time- and dose-dependent manner, which is an important step towards precision medicine. The switch control mechanism of adaptor CARs clearly deviates from current clinical practice to manage CAR T cell-related side effects—e.g., the use of corticosteroids that systemically suppress the entire immune system. Alternative strategies to improve safety—e.g., the integration of suicide genes (inducible Fas or caspase 9, herpes simplex virus thymidine kinase), or elimination genes (CD20, epidermal growth factor receptor (EGFR))—result in CAR T cell depletions that entail the irreversible loss of the costly cell products [19–26]. The controllability of the adaptor CAR therapy will be addressed in Section 3 in more detail.
Due to their dichotomous nature, adaptor CAR platforms overcome the rigid mono-specificity of conventional CARs. This flexibility offers novel possibilities to encounter one of the central problems of conventional CAR T cell therapy—the antigen-negative relapse. One adaptor CAR can redirect T cells against a theoretically unlimited number of target antigens. Thus, the technology allows the manufacturing of one bioengineered T cell product universally applicable for all types of cancer. This obviates the need for the laborious and cost-intensive development of new CAR constructs and genetically modified immune cells. Provided that a comprehensive library of appropriate adaptor molecules is available, adaptor CAR T cells can be easily used for simultaneous or consecutive multiple tumor targeting. This aspect and target specificity of adaptor CAR therapy will be discussed in detail in Section 4.
Since 2006, several groups have devised ten different adaptor CAR platforms that can be subdivided into three major classes (overview Figure 2):
- Fc-binding adaptor CARs;
- Tag-specific adaptor CARs;
- Bispecific antibody (bsAb)-binding adaptor CARs.
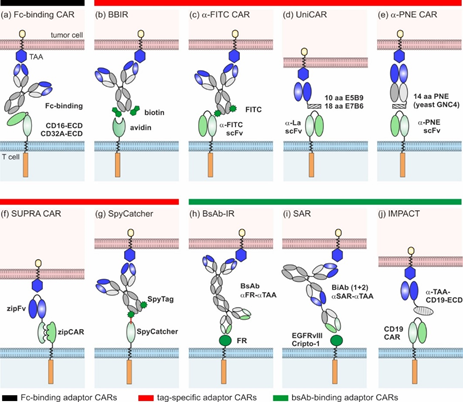
Figure 2. Overview of different adaptor CAR designs: (a) Fc-binding CAR, (b) biotin-binding immune receptors (BBIRs), (c) α-FITC CARs, (d) UniCARs, (e) α-peptide-neoepitope (PNE) CARs, (f) split, universal, and programmable (SUPRA) CARs, (g) SpyCatcher, (h) bispecific antibody-binding immune receptors (BsAb-IRs), (i) synthetic agonistic receptors (SARs), and (j) Integrated Modules oPtimize Adoptive Cell Therapy (IMPACT).
2.1. Fc-Binding Adaptor CARs
Clémenceau and colleagues published the basic idea of adaptor CARs in 2006 [27]. They took advantage of the well-investigated interaction between CD16 and the Fc-part of IgG molecules and constructed the first adaptor CAR composed of the CD16 extracellular domain (ECD) and FceRIg intracellular-signaling domains [27]. Since then, the CD16 adaptor CAR platform was further refined by the construction of different alternative CD16 CARs of the 1st [28,29] and 2nd generation [30,31]. Only in combination with tumor-specific monoclonal antibodies (mAb) (e.g., rituximab, trastuzumab, cetuximab), CD16 CAR T cells triggered efficient tumor lysis both in vitro and in vivo (Figure 2a) [27–31]. Due to the low binding affinity of CD16 to human IgG2, Caratelli et al. later utilized the CD32A ECD for the construction of adaptor CARs that are able to bind IgG1 and IgG2 with similar affinities [32]. The therapeutic activity of antibodies is clearly determined by their glycosylation pattern [33]. Accordingly, glyco-engineered antibodies were shown to amplify CD16 CAR T cell activity [31]. As this might entail an increased risk of severe side effects (e.g., CRS), suitable adaptor mAbs need to be assessed carefully [31]. A major advantage of Fc-binding adaptor CAR systems is the availability of a wide repertoire of antibody-dependent cellular cytotoxicity (ADCC)-mediating mAbs in clinical grade quality [34], circumventing extensive antibody engineering and easily enabling target switch during therapy. Although therapeutic effects might be hampered by interference with naturally occurring IgG molecules, it was shown that an excess of irrelevant immunoglobulins did not inhibit but rather increased the cytotoxic capacity of the CD16 adaptor CAR systems [30,31]. The authors assume that the unspecific deposition of IgG on cancer cells was responsible for enhanced killing and thus recommend excluding patients with IgG-associated pathologies from CD16 CAR therapy [31].
2.2. Tag-Binding Adaptor CARs
In general, tag-specific adaptor CARs harbor an ECD that recognizes a chemically, enzymatically, or genetically attached tag of tumor-specific adaptor molecules.
Biotin-binding immune receptor (BBIR) T cells were the first tag-specific adaptor CARs described in the literature (Figure 2b) [35]. They harness the highly specific, non-covalent interaction of avidin or streptavidin with biotin. The 67 kDa chicken avidin and the 53 kDa bacterial streptavidin (Streptomyces avidinii) can bind up to four biotin molecules simultaneously. Based on these proteins, different BBIRs were designed, carrying either monomeric or dimeric biotin-binding molecules as ECDs [35,36]. In combination with biotinylated mAbs or single-chain fragment variables (scFvs), only dimeric chicken avidin (dcAv) CAR T cells and monomeric streptavidin (mSA2) CAR T cells were proven to be useful tools for the in vitro and in vivo targeting of various cancer cells overexpressing, e.g., epithelial cell adhesion molecule (EpCAM), EGFR, and CD20 [35,36]. As soluble biotin did not inhibit the performance of BBIR CAR T cells, the risk of interference with biotin naturally present in patients seems to be low [35,37]. However, the occurrence of natural anti-biotin antibodies in human serum [38] and the antigenicity of avidin and streptavidin (e.g., [39,40]) might interfere with therapeutic effects. Whether this can induce undesirable adverse reactions or hamper the successful clinical translation of the BBIR adaptor system has yet to be investigated.
Another semi-synthetic adaptor CAR system relies on scFv-based α-FITC CARs targeting the synthetic dye fluorescein isothiocyanate (FITC) that is chemically coupled to various tumor-specific adaptor molecules (Figure 2c) [41]. The first proof of concept studies by Tamada and colleagues demonstrated that T cells carrying 3rd generation α-FITC CARs are able to elicit potent anti-tumor responses in the presence of FITC-labeled cetuximab, rituximab, and trastuzumab [41–49]. Over time, different classes of FITC-conjugated adaptor molecules were studied, including targeting compounds based on fragments of antigen binding (Fabs) [43,44] and small molecules [42,45–49]. As shown for HER2- and CD19- and CD20-Fabs, the position and stoichiometry of the FITC label influenced the α-FITC CAR T cell activity [43,44]. The consequential need for the individual optimization of each target contradicts the fast adaptability of adaptor CAR platforms. The most widely studied adaptor molecule is EC17, a folate-FITC conjugate [42,46–49] initially designed and clinically tested (ClinicalTrials.gov. Identifier: NCT01996072, NCT01994369, NCT02000778, NCT01778933, NCT01778920, NCT01511055, NCT00485563) for the image-guided surgery of inoperative solid tumors (e.g., [50]). The small molecular weight adaptor efficiently redirected the α-FITC CAR T cells against different tumor entities—e.g., non-small lung cancer, breast cancer, and osteosarcoma—allowing for controllability in a time- and dose-dependent manner [46–49]. Despite the full humanization of the α‑FITC CAR T cell product [44], the immunogenic potential of FITC is one concern for clinical translation, as underlined by the emergence of α-FITC antibodies in therapeutic mouse models [41].
Alternatively, adaptor molecules can be endowed with small peptide tags to redirect standard scFv-based adaptor CARs. The UniCAR platform introduced in 2014 [51,52] utilizes the 10 amino acid (aa) peptide epitope E5B9 derived from the human nuclear La/SS-B protein [53,54]. Meanwhile, a broad library of E5B9-tagged adaptor molecules, so-called target modules (TMs), were developed to specifically cross-link UniCAR T cells with tumor cells (Figure 2d). They were built on different binding moieties (small peptide molecules, nanobodies, and scFvs), targeting various antigens overexpressed in hematologic and solid tumors (e.g., CD19, CD33, CD123, CD98, EGFR, disialoganglioside (GD2), prostate stem cell antigen (PSCA), prostate-specific membrane antigen (PSMA), and sialyl-Tn (STn)) [52,55–63]. Recently, larger IgG-based TMs were effectively used for the redirection of UniCAR T cells against GD2- and STn-expressing cancer cells in vitro and in vivo [64,65]. The anti-tumor responses were comparable to small scFv-based TMs, but the serum half-lives considerably increased, which will impact future dosing regimens (see Section 3). Apart from E5B9, the 18 aa α-helical La epitope E7B6 was also successfully employed in the UniCAR system [66]. Although La/SS-B is a well-known autoantigen, the immunogenic potential of E5B9 and E7B6 is expected to be very low, as both epitopes are cryptic in the native La protein and none of the La-specific antibodies found in the sera of >100 autoimmune patients showed reactivity against these epitopes [67–71]. To reduce the overall immunogenicity of UniCAR components, the α-La and tumor-specific scFvs of UniCARs and TMs were humanized, respectively. Similarly to the UniCAR approach, scFv-based α-peptide neo-epitope (PNE) CARs recognize adaptor molecules endowed with a 14 aa peptide epitope derived from the yeast transcription factor GNC4 (Figure 2e) [43,72–74]. As this PNE is not naturally occurring in humans, it is at greater risk of inducing immune responses in patients. All the PNE-tagged adaptor molecules described so far were constructed based on tumor-specific IgG molecules or Fabs, while the latter were preferred due to their favorable pharmacokinetic properties [72]. PNE-tagged Fabs against CD19, CD20, and HER2 effectively redirected adaptor CAR T cells for the lysis of B cell lines [72] and breast cancer cell lines [43], as well as patient-derived pancreatic cancer cells [73]. The α-PNE CAR T cell activity and phenotype was temporarily controlled via adaptor molecule dosing [72,74]. Interestingly, the modifications of α-PNE CAR hinge region, PNE conjugation site, and number considerably altered the geometry of the immunological synapses and thereby influenced the overall performance of the α‑PNE adaptor CAR T cells [72]. The best adaptor molecule designs were dependent on the selected tumor antigen [43,72], as was also observed for FITC-labeled Fab-switches [43,44].
In 2018, Cho et al. introduced the split, universal, and programmable (SUPRA) CAR technology (Figure 2f), which utilizes leucine zippers as interaction partners between adaptor CAR T cells and adaptor molecules [75]. The so-called zipCAR was designed by the fusion of a leucine zipper to the intracellular signaling domains of 4-1BB and CD3z. ZipFvs function as adaptor molecules and are composed of tumor-specific scFvs (α-HER2, α-AXL, α-mesothelin) and a cognate leucine zipper. The amphipathic interactions between two ZIP domains determine the zipCAR/zipFv affinities that are exploited to fine-tune the specificity and activity of zipCAR T cells (see Sections 3 and 4). In addition to synthetic leucine zippers, leucine zippers derived from human FOS and JUN were used to create a humanized SUPRA CAR system.
SpyCatcher immune receptors are a novel class of adaptor CARs. Unlike others, they are able to covalently bind SpyTag-containing adaptor molecules (Figure 2g) [76]. The SpyTag/SpyCatcher system originated from the immunoglobulin-like collagen adhesion domain of Streptococcus pyogenes (CnaB2), which contains an internal isopeptide bond between aa position 31 (Lys) and 117 (Asp) [77]. The separation of CnaB2 and subsequent modifications resulted in the Lys31-containing SpyTag peptide (13 aa) and Asp117-containing SpyCatcher protein (116 aa) [77]. Both binding partners first associate non-covalently with a high affinity, rapidly followed by a spontaneous, autocatalytic isopeptide bond formation between Lys31 and Asp117 [77]. To create 2nd generation SpyCatcher CARs, the SpyCatcher protein was connected with the intracellular CD3z and CD28 or 4-1BB signaling domains [76]. The SpyTag in turn was genetically fused or site-specifically attached to HER2-, EGFR-, EpCAM-specific Designed Ankyrin Repeat Proteins (DARPins), and clinical-grade IgG molecules (rituximab, trastuzumab, cetuximab) [76]. In the first proof of concept studies, Minotulu et al. [76] demonstrated that SpyTag-containing adaptor molecules were efficiently attached to SpyCatcher-immune receptor-equipped T cells and subsequently mediated efficient tumor cell lysis in vitro and in vivo. Upon antigen-specific stimulation, preloaded SpyCatcher immune receptors are internalized, ensuring an off-switch. Thus, adaptor CAR T cells lose their target specificity over time and require continued rearming with SpyTag-containing adaptor molecules. The possibility of covalently arming SpyCatcher adaptor CAR T cells with one or multiple target specificities prior to infusion is a unique feature of this system.
2.3. BsAb-Binding Adaptor CARs
Due to their dual specificity for a tumor-specific antigen and an activating immune receptor (e.g., CD3), bsAbs are able to redirect T cells for highly efficient tumor cell killing [78]. In 2014, Urbanska and colleagues conceived the idea to combine the power of bsAbs with CARs; they developed the first bsAb-binding immune receptor (bsAb-IR), comprising the extracellular part of human folate receptor α (FRα) (231 aa) and 1st or 2nd generation CAR signaling domains (Figure 2h) [79]. Bispecific adaptor molecules were created by the chemical heteroconjugation of α-FRα and α-CD20 mAbs. Although this was the first in vitro data to verify the general functionality of the system, the lytic activity of the redirected FRα CAR T cells against B cell lines was low due to the poor quality of the bsAb adaptors [79]. Later, Karches et al. presented alternative bsAb-binding adaptor CARs containing the ECD of human epidermal growth factor receptor variant III (EGFRvIII) or human Cripto-1 and termed them synthetic agonistic receptors (SARs) (Figure 2i) [80]. In their studies, they explored both tetravalent (2 + 2) and trivalent (2 + 1) bispecific adaptors targeting EpCAM or mesothelin in murine and human mouse models. Data have proven that only bsAbs with one binding arm for the SAR-ECD are able to trigger T cell activation, proliferation, and tumor lysis in a strictly target-dependent manner. To avoid cross-reactivity with healthy tissues, the ECD of bsAb-binding adaptor CARs should be carefully selected. As EGFRvIII is exclusively expressed in malignant cells and Cripto-1 is an embryonic antigen, they possess a relatively low risk of unwanted side effects.
In 2017, Ambrose and colleagues introduced the IMPACT (Integrated Modules oPtimize Adoptive Cell Therapy) strategy (Figure 2j) [81,82]. They refashioned conventional CD19 CAR T cells into adaptor CARs. Bifunctional fusion proteins, which are composed of an optimized variant of the CD19-ECD and a tumor-specific binding moiety (e.g., scFv), served as bridging molecules between the tumor and CD19 CAR T cells [81–83]. By using this approach, the CD19 CAR T cells elicited potent anti-tumor responses in experimental mice models—e.g., against CD19negHER2pos and CD19negCD20pos tumors. The adaptor molecules were delivered either via infusion or directly via CD19 CAR T cells [81–83]. To achieve the latter, lentiviral constructs encoding the CD19 CAR and the adaptor molecule were designed. Overall, the IMPACT strategy is a promising method to repurpose CD19 CAR T cells for targeting alternative tumor-associated antigens after CD19neg disease relapse, such as CD20 [83]. Although the versatility of the systems was proven for solid tumors [82], the risk of CD19 CAR T cell therapy-related side effects such as CRS, CRES, and B cell aplasia remains and impairs the safety profile of this approach.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12051302