Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Major depressive disorder (MDD) is a heterogeneous neuropsychological disorder characterized by a combination of symptoms that negatively impact the productivity and well-being of inflicted patients, including impairments in cognition, emotional regulation, memory, motor function, motivation, and possible suicidal ideation. Glutamate is the major excitatory neurotransmitter in the central nervous system. It plays an important role in several physiological functions.
- glutamate
- AMPA
- NMDA
- depression
- chronic stress
1. Introduction
Major depressive disorder (MDD) is a heterogeneous neuropsychological disorder characterized by a combination of symptoms that negatively impact the productivity and well-being of inflicted patients, including impairments in cognition, emotional regulation, memory, motor function, motivation, and possible suicidal ideation. Approximately 280 million people in the world are affected by this psychiatric disorder, and it is regarded as one of the leading causes of morbidity and disability worldwide [1]. An episode of major depression may occur once in a person’s lifetime, but it is more likely to relapse throughout a person’s life [2]. Although several pharmaceutical strategies have been proven in clinical settings, more than two-thirds of patients with MDD do not achieve stable remission of symptoms, despite currently available medication [3]. Furthermore, the current antidepressant drugs targeting the monoamine systems (norepinephrine, dopamine, and/or serotonin) require a long duration (4–8 weeks) to take effect [4]. The severe adverse effects associated with the pharmacotherapy, including cardiac toxicity, hyperpiesia, sexual dysfunction, body weight gain, and sleep disorders, often hamper patients’ adherence [5][6]. The current pharmacotherapy for MDD remains unsatisfactory, which may be attributed to the lack of understanding of the neurobiological mechanisms underlying this pathological condition.
MDD is a debilitating disorder with multiple potential etiologies, including environmental and genetical influences [7]. Although several hypotheses have been suggested to explain the pathophysiological mechanisms of MDD, the contours of the disorder are still unclear. Among these hypotheses, the dysregulation of neurotransmitters has received the most attention in the neurobiological mechanisms of MDD, i.e., the monoaminergic hypothesis involving serotonin, norepinephrine, and/or dopamine. Clinically, most antidepressants target this system as a therapeutic goal, including tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), serotonin, and norepinephrine reuptake inhibitors (SNRIs), to restore the levels of these monoamines [8].
A growing consensus posits that altered monoaminergic transmission is insufficient to explain the etiology of depressive disorders [9][10][11]. Hypothalamic–pituitary–adrenal (HPA) axis dysregulation has been implicated in the pathogenesis of MDD, as the hyperactivity state of the HPA axis is one of the most consistent findings in the neuroendocrinology of depression [12][13] and as the HPA axis is dysregulated by stress and trauma, both of which are known precipitants of MDD [14]. Furthermore, data from rodents showed that central administration of corticosterone, a stress-related hormone, can produce depression-like behaviors [15]. The plasma levels of corticosterone, a stress hormone, have been widely used as biomarkers of stress [16], depression [17] and anxiety [18] in mice, and such a phenomenon was also observed in humans [19] in the form of plasma cortisol. Chronic stress is reported to induce neuropsychological disorders, including depression and anxiety, via the hyperactivity of the hypothalamic–pituitary–adrenal (HPA) axis [20][21].
In addition to the monoaminergic system, a growing body of evidence has revealed that the glutamatergic system plays a critical role in the pathophysiological mechanisms of MDD [1][22][23]. Several postmortem studies have indicated that glutamate receptor subunits, including the GluR1 subunit and AMPA binding, were reduced in patients with major depression [22][23][24][25]. Beyond the conventional monoaminergic theory, these findings provided an alternative concept in the possible pathological mechanisms of MDD and the different approach in the development of novel antidepressants.
2. A Brief Overview of Glutamate Neurotransmission in the Brain
Glutamate is the major excitatory neurotransmitter in the central nervous system. It plays an important role in several physiological functions. A typical glutamate synapse is composed of three distinct cell types: an astrocyte, a presynaptic neuron, and a postsynaptic neuron [26]. In the central nervous system, glutamine synthesis from glutamate and ammonia occurs exclusively in the astrocytes. Then, glutaminase, a precursor for the synthesis of the neurotransmitter glutamate, hydrolyses glutamine to glutamate and ammonia within neurons, completing the glutamate–glutamine cycle in the brain (Figure 1). Glutamate is then packaged into vesicles in the presynaptic neuron by three types of vesicular glutamate transporters (VGLUT1, VGLUT2 and VGLUT3) and released into the synapse from the presynaptic terminals [27], in an activity-dependent manner. The released glutamate in the synaptic cleft can engage both ionotropic and metabotropic glutamate receptors, located in the presynaptic and postsynaptic neurons, as well as on astrocytes [28]. The ionotropic subtypes of glutamate receptors, including the AMPA, N-methyl-D-aspartate (NMDA), and kainate receptors, control fast excitatory neurotransmission, whereas the family of eight metabotropic glutamate receptors (mGluR1-8) are located in presynaptic areas, postsynaptic areas, and extra-synaptically throughout the central nervous system [29]. The concentrations of synaptic glutamate are regulated by glutamate transporters localized in glial cells and neurons due to the fact that there is no process by which glutamate is metabolized in neurons.
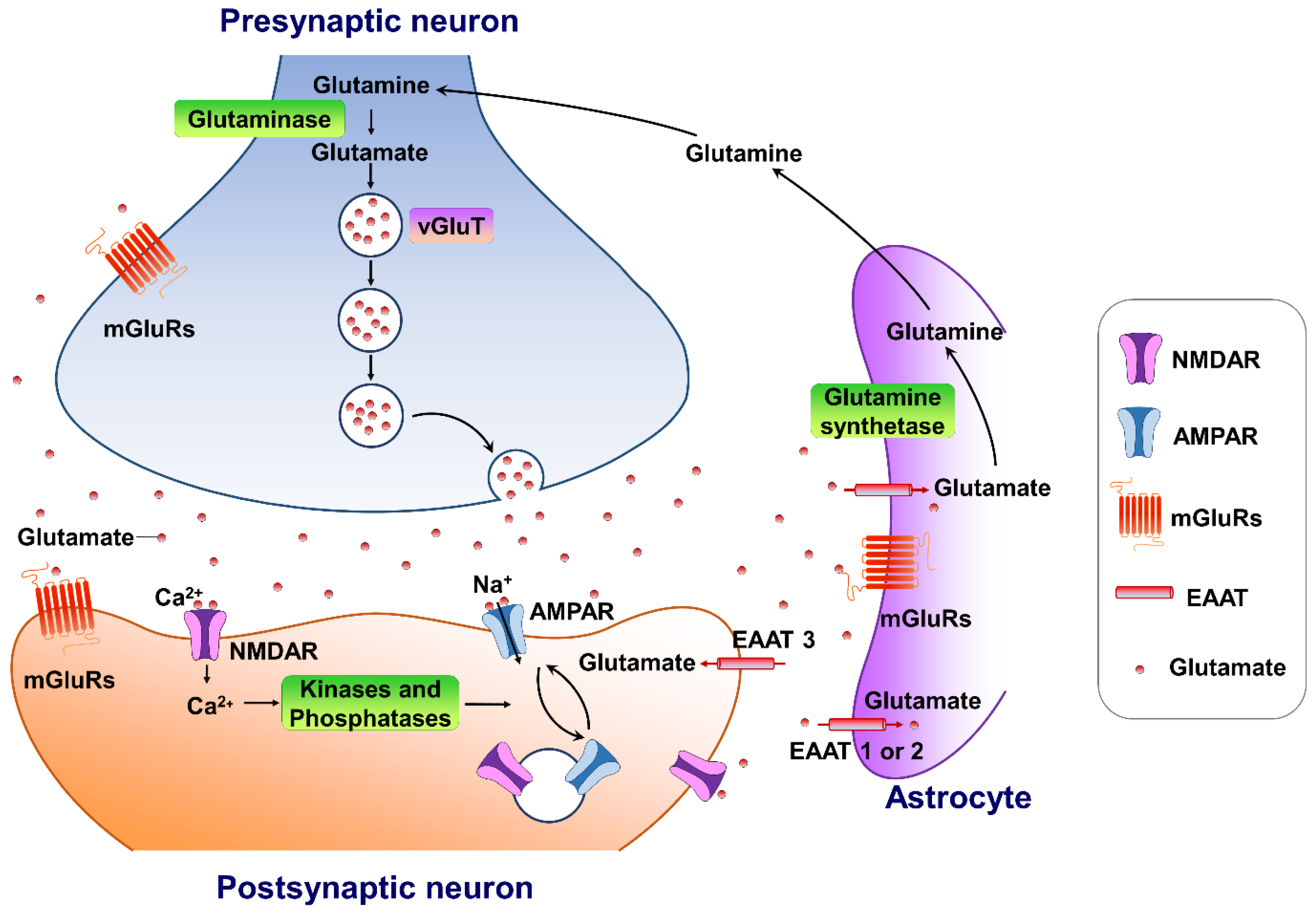 Figure 1. The tripartite glutamate synapses. Most glutamate molecules are cleared from the synaptic cleft through the excitatory amino-acid transporter (EAAT 1/2) located on the astrocytes. Within the astrocyte, glutamine synthetase converts glutamate to glutamine, and the glutamine is subsequently released from the astrocyte and taken up by neighboring neurons to complete the glutamate–glutamine cycle in the brain. Neuronal glutamate is synthesized de novo from glutamine originating from nearby astrocytes. Glutamate is then loaded into synaptic vesicles by vesicular glutamate transporters (VGLUTs). Upon being triggered by an action potential, glutamate will be rapidly released into the synaptic cleft. Here, glutamate binds to either ionotropic glutamate receptors (AMPA receptors and NMDA receptors) and/or metabotropic glutamate receptors (mGluRs) on the membranes of both postsynaptic and presynaptic neurons and astrocytes. Upon activation, these glutamate receptors initiate various cellular responses, including depolarization of membrane potential, activation of intracellular signaling, regulation of protein synthesis, and/or gene expression. Surface expression and functional alteration of AMPARs and NMDARs are dynamically mediated by protein synthesis and degradation. The receptors traffic between the postsynaptic membrane and endosomes to maintain the dynamic adaptation/alteration of physiological/pathological responses.
Figure 1. The tripartite glutamate synapses. Most glutamate molecules are cleared from the synaptic cleft through the excitatory amino-acid transporter (EAAT 1/2) located on the astrocytes. Within the astrocyte, glutamine synthetase converts glutamate to glutamine, and the glutamine is subsequently released from the astrocyte and taken up by neighboring neurons to complete the glutamate–glutamine cycle in the brain. Neuronal glutamate is synthesized de novo from glutamine originating from nearby astrocytes. Glutamate is then loaded into synaptic vesicles by vesicular glutamate transporters (VGLUTs). Upon being triggered by an action potential, glutamate will be rapidly released into the synaptic cleft. Here, glutamate binds to either ionotropic glutamate receptors (AMPA receptors and NMDA receptors) and/or metabotropic glutamate receptors (mGluRs) on the membranes of both postsynaptic and presynaptic neurons and astrocytes. Upon activation, these glutamate receptors initiate various cellular responses, including depolarization of membrane potential, activation of intracellular signaling, regulation of protein synthesis, and/or gene expression. Surface expression and functional alteration of AMPARs and NMDARs are dynamically mediated by protein synthesis and degradation. The receptors traffic between the postsynaptic membrane and endosomes to maintain the dynamic adaptation/alteration of physiological/pathological responses.3. Targeting AMPA-Glutamatergic Signaling for the Development of Novel Therapeutics for Depressive Disorders
Emerging evidence of glutamatergic synaptic transmission dysfunctions demonstrates how this defect correlates with the pathogenesis of depression. MDD is attributed to a weakening of specific subsets of excitatory synapses in various brain nuclei that are important in the processing of affect and reward (Table 1). The critical action of effective antidepressants is the reversal of the diminished excitatory synaptic strength in these impaired brain areas. A candidate agent verified in preclinical models that promotes stress-sensitive excitatory synapses in the emotional and reward circuits may be an effective antidepressant. Reduced glutamate levels have been noted in several neural regions of patients with MDD [30], whereas several glutamatergic agents have been proven to effectively alleviate depressive symptoms in patients with MDD [31]. Accumulating evidence demonstrates that transcriptional regulation is involved in various levels of regulation of gene expression [32]. Chronic stress causes a loss of AMPA receptors, and GluR1 expression in the PFC is attributed to the enhancement of ubiquitin–proteasome-dependent degradation of GluR1 controlling by E3 ubiquitin ligase [33]. Transcriptional regulation of gene expression has been considered as a key player underlying the persistent impact of stress response.
Table 1. Effects of AMPA receptor response to different stress paradigms. ↑ = increase; ↓ = decrease; - = no change.
| Classification | Type of Stress | Brain Area | Effects on AMPA Receptor | Reference |
|---|---|---|---|---|
| Acute stress | Restraint stress for 2 h | Hippocampus | GluA1 subunit phosphorylation ↑ | [34] |
| Basolateral amygdala | GluA1 subunit phosphorylation - | [34] | ||
| Frontal cortex | GluA1 subunit phosphorylation - | [34] | ||
| Restraint stress for 30 min | Hippocampus | GluA1 subunit phosphorylation ↑ GluA1 expression ↑ |
[35] | |
| Unsteady platform for acute stress | Hippocampus | GluA1 expression ↓ | [36] | |
| Acute footshock stress | Prefrontal and frontal cortex | GluA1 subunit phosphorylation ↑ GluA2 subunit phosphorylation ↑ |
[37] | |
| Elevated platform stress | Amygdala | GluA1 subunit phosphorylation ↑ | [38] | |
| mPFC | GluA1 subunit phosphorylation ↑ | [38] | ||
| Hippocampus | GluA1 subunit phosphorylation ↑ | [38] | ||
| Acute restraint stress for 1 h | Hippocampus | GluA1 expression - GluA2 expression - |
[39] | |
| Acute restraint stress for 30 min | Hippocampus | GluA1 subunit phosphorylation ↑ | [40] | |
| Elevated platform stress | Hippocampus | GluA2 expression ↓ | [41] | |
| Restraint or forced swimming | Amygdala | GluA1 subunit phosphorylation ↑GluA1 expression ↑ | [42] | |
| Acute restraint stress for 2 h | Nucleus accumbens | GluA1 expression ↑ | [43] | |
| Unsteady platform for 30 min | mPFC | Ser831-GluA1 phosphorylation ↓ Ser880-GluA2 phosphorylation ↑ |
[44] | |
| Hippocampus | Ser831-GluA1 phosphorylation ↓ | [44] | ||
| Amygdala | Ser845-GluA1 phosphorylation ↑ Tyr876-GluA2 phosphorylation ↓ Ser880-GluA2 phosphorylation ↓ |
[44] | ||
| Forced-swim stress | Prefrontal cortex | Surface GluA1 expression ↑ Surface GluA2 expression ↑ |
[45] | |
| Immobilization stress for 45 min | Hippocampus | AMPA mRNA levels - | [46] | |
| Acute restraint stress for 6 h | Dentate gyrus | GluR2 flip mRNA expression↑ | [47] | |
| Chronic stress | Chronic mild stress | Hippocampus | AMPA mRNA - GluA2 expression ↑ |
[48] |
| Chronic unpredictable mild stress | Hippocampus | GluA1 expression - GluA2 expression↑ GluA3 expression ↑ |
[49] | |
| Early life Stress | Hippocampus | NMDA/AMPA ratio ↓ | [50] | |
| Chronic unpredictable mild stress | Hippocampus | GluA1 expression ↓ GluA2 expression ↓ GluA1 subunit phosphorylation ↓ |
[51] | |
| Week chronic mild stress | Hippocampus | GluA1 expression ↓ | [52] | |
| Chronic unpredictable stress | Hippocampus | GluA1 expression ↓ | [53] | |
| Neonatal isolation stress | Paraventricular nucleus | AMPA binding sites ↑ | [54] | |
| Chronic immobilized stress | Nucleus accumbens | GluA1 expression ↑ | [55] | |
| Immobilization stress for 14 days | Hippocampus | AMPA mRNA levels - | [46] | |
| Chronic restraint stress for 21 days | Hippocampus CA1 | GluR1 flip mRNA expression ↓ | [47] |
Despite mounting positive preclinical results, most AMPA receptor-targeting agents have failed to declare any relevant clinical effects in treating neuropsychiatric disorders. This discrepancy may be due to multiple factors. Nonetheless, strong preclinical evidence supports that AMPA receptors are involved in the therapeutic effects of current antidepressant drugs. Chronic restraint stress-induced depression in rodents was reversed by the metabolite of ketamine, (2R,6R)-HNK, via AMPA receptors [56] at the concentration that do not block NMDA receptor function [57]. On the other hand, mPFC AMPA receptor and BDNF signaling are required for the rapid and sustained antidepressant-like effects of 5-HT1A receptor stimulation [58]. Interestingly, pharmacological enhancement of AMPA transmission has been reported to synergize the antidepressant potency of conventional antidepressants, e.g., fluoxetine, imipramine, and rolipram [59]. Given that AMPA-glutamatergic neurotransmission in various stress-related brain regions signifies the dysfunction of excitatory synapse, it could point to a new strategy for developing novel antidepressants or as a target for adjunct therapy alongside current antidepressants (Figure 2).
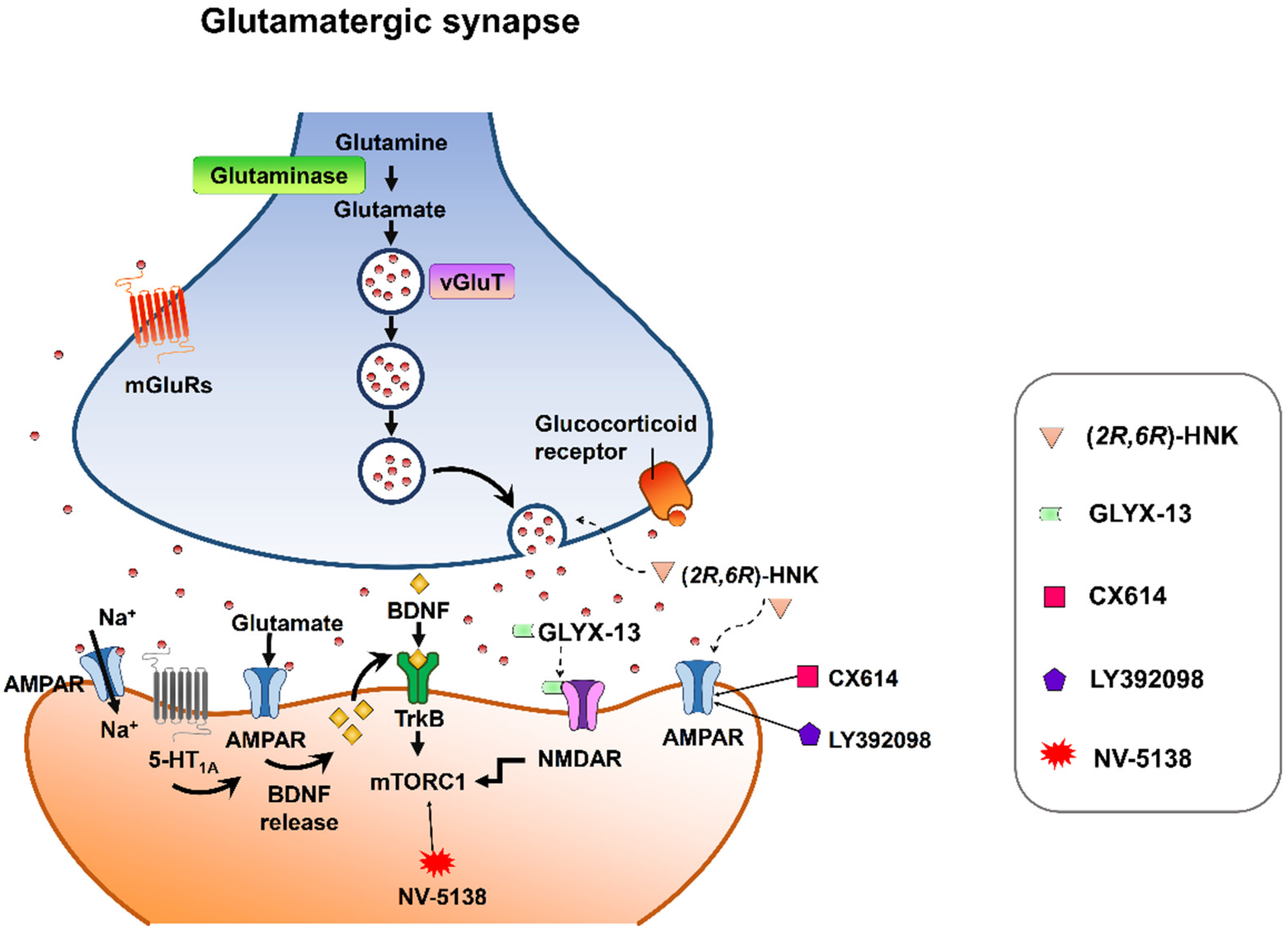 Figure 2. Synaptic model for the cellular target sites for different types of candidate drugs for antidepressants. (2R,6R)-HNK exerts increased glutamate release and AMPA receptor-mediated synaptic potentiation. GLYX-13 elicited partial activation of the NMDA receptor, hence activation of mTORC1 and thus induction of protein synthesis. CX614 and LY392098, AMPA receptor potentiators, induce antidepressant effects by enhancement of AMPA receptor function and BDNF expression. NV-5138 exerts rapid and sustained antidepressant effects through stimulating mTORC1 signaling. Activation of the 5-HT1A receptor produces rapid and sustained antidepressant effects through the initiation of AMPA receptor/BDNF/mTORC1 cascades. All the candidates propose long-lasting modifications in synaptic plasticity, resulting in strengthening of glutamatergic synapses, which is necessary for antidepressant responses.
Figure 2. Synaptic model for the cellular target sites for different types of candidate drugs for antidepressants. (2R,6R)-HNK exerts increased glutamate release and AMPA receptor-mediated synaptic potentiation. GLYX-13 elicited partial activation of the NMDA receptor, hence activation of mTORC1 and thus induction of protein synthesis. CX614 and LY392098, AMPA receptor potentiators, induce antidepressant effects by enhancement of AMPA receptor function and BDNF expression. NV-5138 exerts rapid and sustained antidepressant effects through stimulating mTORC1 signaling. Activation of the 5-HT1A receptor produces rapid and sustained antidepressant effects through the initiation of AMPA receptor/BDNF/mTORC1 cascades. All the candidates propose long-lasting modifications in synaptic plasticity, resulting in strengthening of glutamatergic synapses, which is necessary for antidepressant responses.This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10051005
References
- Pinto, B.; Conde, T.; Domingues, I.; Domingues, M.R. Adaptation of Lipid Profiling in Depression Disease and Treatment: A Critical Review. Int. J. Mol. Sci. 2022, 23, 2032.
- Belmaker, R.H.; Agam, G. Major depressive disorder. N. Engl. J. Med. 2008, 358, 55–68.
- Li, G.; Fife, D.; Wang, G.; Sheehan, J.J.; Boden, R.; Brandt, L.; Brenner, P.; Reutfors, J.; DiBernardo, A. All-cause mortality in patients with treatment-resistant depression: A cohort study in the US population. Ann. Gen. Psychiatry 2019, 18, 23.
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. Lancet 2018, 391, 1357–1366.
- Ho, S.C.; Jacob, S.A.; Tangiisuran, B. Barriers and facilitators of adherence to antidepressants among outpatients with major depressive disorder: A qualitative study. PLoS ONE 2017, 12, e0179290.
- Hung, C.I. Factors predicting adherence to antidepressant treatment. Curr. Opin. Psychiatry 2014, 27, 344–349.
- Jesulola, E.; Micalos, P.; Baguley, I.J. Understanding the pathophysiology of depression: From monoamines to the neurogenesis hypothesis model—Are we there yet? Behav. Brain Res. 2018, 341, 79–90.
- Manji, H.K.; Drevets, W.C.; Charney, D.S. The cellular neurobiology of depression. Nat. Med. 2001, 7, 541–547.
- Heninger, G.R.; Delgado, P.L.; Charney, D.S. The revised monoamine theory of depression: A modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry 1996, 29, 2–11.
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61 (Suppl. 6), 4–6.
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12.
- Nestler, E.J.; Barrot, M.; DiLeone, R.J.; Eisch, A.J.; Gold, S.J.; Monteggia, L.M. Neurobiology of depression. Neuron 2002, 34, 13–25.
- Iob, E.; Kirschbaum, C.; Steptoe, A. Persistent depressive symptoms, HPA-axis hyperactivity, and inflammation: The role of cognitive-affective and somatic symptoms. Mol. Psychiatry 2020, 25, 1130–1140.
- Heim, C.; Newport, D.J.; Wagner, D.; Wilcox, M.M.; Miller, A.H.; Nemeroff, C.B. The role of early adverse experience and adulthood stress in the prediction of neuroendocrine stress reactivity in women: A multiple regression analysis. Depress. Anxiety 2002, 15, 117–125.
- Skorzewska, A.; Lehner, M.; Wislowska-Stanek, A.; Krzascik, P.; Ziemba, A.; Plaznik, A. The effect of chronic administration of corticosterone on anxiety- and depression-like behavior and the expression of GABA-A receptor alpha-2 subunits in brain structures of low- and high-anxiety rats. Horm. Behav. 2014, 65, 6–13.
- Kim, J.G.; Jung, H.S.; Kim, K.J.; Min, S.S.; Yoon, B.J. Basal blood corticosterone level is correlated with susceptibility to chronic restraint stress in mice. Neurosci. Lett. 2013, 555, 137–142.
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006.
- McGill, B.E.; Bundle, S.F.; Yaylaoglu, M.B.; Carson, J.P.; Thaller, C.; Zoghbi, H.Y. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 18267–18272.
- Nandam, L.S.; Brazel, M.; Zhou, M.; Jhaveri, D.J. Cortisol and Major Depressive Disorder-Translating Findings From Humans to Animal Models and Back. Front. Psychiatry 2019, 10, 974.
- Faravelli, C.; Lo Sauro, C.; Godini, L.; Lelli, L.; Benni, L.; Pietrini, F.; Lazzeretti, L.; Talamba, G.A.; Fioravanti, G.; Ricca, V. Childhood stressful events, HPA axis and anxiety disorders. World J. Psychiatry 2012, 2, 13–25.
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M., Jr.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 2017, 22, 527–536.
- Choudary, P.V.; Molnar, M.; Evans, S.J.; Tomita, H.; Li, J.Z.; Vawter, M.P.; Myers, R.M.; Bunney, W.E., Jr.; Akil, H.; Watson, S.J.; et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. USA 2005, 102, 15653–15658.
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902.
- Freed, W.J.; Dillon-Carter, O.; Kleinman, J.E. Properties of AMPA binding in postmortem human brain from psychotic subjects and controls: Increases in caudate nucleus associated with suicide. Exp. Neurol. 1993, 121, 48–56.
- Meador-Woodruff, J.H.; Hogg, A.J., Jr.; Smith, R.E. Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Res. Bull. 2001, 55, 631–640.
- Zarate, C.A.; Quiroz, J.; Payne, J.; Manji, H.K. Modulators of the glutamatergic system: Implications for the development of improved therapeutics in mood disorders. Psychopharmacol. Bull. 2002, 36, 35–83.
- Takamori, S.; Rhee, J.S.; Rosenmund, C.; Jahn, R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 2000, 407, 189–194.
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105.
- Watkins, J.C.; Jane, D.E. The glutamate story. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S100–S108.
- Arnone, D.; Mumuni, A.N.; Jauhar, S.; Condon, B.; Cavanagh, J. Indirect evidence of selective glial involvement in glutamate-based mechanisms of mood regulation in depression: Meta-analysis of absolute prefrontal neuro-metabolic concentrations. Eur. Neuropsychopharmacol. 2015, 25, 1109–1117.
- Henter, I.D.; de Sousa, R.T.; Zarate, C.A., Jr. Glutamatergic Modulators in Depression. Harv. Rev. Psychiatry 2018, 26, 307–319.
- Seeburg, P.H. A-to-I editing: New and old sites, functions and speculations. Neuron 2002, 35, 17–20.
- Wei, J.; Yuen, E.Y.; Liu, W.; Li, X.; Zhong, P.; Karatsoreos, I.N.; McEwen, B.S.; Yan, Z. Estrogen protects against the detrimental effects of repeated stress on glutamatergic transmission and cognition. Mol. Psychiatry 2014, 19, 588–598.
- Novaes, L.S.; Dos Santos, N.B.; Perfetto, J.G.; Goosens, K.A.; Munhoz, C.D. Environmental enrichment prevents acute restraint stress-induced anxiety-related behavior but not changes in basolateral amygdala spine density. Psychoneuroendocrinology 2018, 98, 6–10.
- Wang, M.; Ramasamy, V.S.; Samidurai, M.; Jo, J. Acute restraint stress reverses impaired LTP in the hippocampal CA1 region in mouse models of Alzheimer’s disease. Sci. Rep. 2019, 9, 10955.
- Li, C.; Zhang, J.; Xu, H.; Chang, M.; Lv, C.; Xue, W.; Song, Z.; Zhang, L.; Zhang, X.; Tian, X. Retigabine ameliorates acute stress-induced impairment of spatial memory retrieval through regulating USP2 signaling pathways in hippocampal CA1 area. Neuropharmacology 2018, 135, 151–162.
- Bonini, D.; Mora, C.; Tornese, P.; Sala, N.; Filippini, A.; La Via, L.; Milanese, M.; Calza, S.; Bonanno, G.; Racagni, G.; et al. Acute Footshock Stress Induces Time-Dependent Modifications of AMPA/NMDA Protein Expression and AMPA Phosphorylation. Neural Plast. 2016, 2016, 7267865.
- Caudal, D.; Rame, M.; Jay, T.M.; Godsil, B.P. Dynamic Regulation of AMPAR Phosphorylation In Vivo Following Acute Behavioral Stress. Cell. Mol. Neurobiol. 2016, 36, 1331–1342.
- Jin, Y.; Kanno, T.; Nishizaki, T. Acute restraint stress impairs induction of long-term potentiation by activating GSK-3beta. Neurochem. Res. 2015, 40, 36–40.
- Whitehead, G.; Jo, J.; Hogg, E.L.; Piers, T.; Kim, D.H.; Seaton, G.; Seok, H.; Bru-Mercier, G.; Son, G.H.; Regan, P.; et al. Acute stress causes rapid synaptic insertion of Ca2+ -permeable AMPA receptors to facilitate long-term potentiation in the hippocampus. Brain 2013, 136, 3753–3765.
- Sebastian, V.; Estil, J.B.; Chen, D.; Schrott, L.M.; Serrano, P.A. Acute physiological stress promotes clustering of synaptic markers and alters spine morphology in the hippocampus. PLoS ONE 2013, 8, e79077.
- Tian, Z.; Wang, Y.; Zhang, N.; Guo, Y.Y.; Feng, B.; Liu, S.B.; Zhao, M.G. Estrogen receptor GPR30 exerts anxiolytic effects by maintaining the balance between GABAergic and glutamatergic transmission in the basolateral amygdala of ovariectomized mice after stress. Psychoneuroendocrinology 2013, 38, 2218–2233.
- Garcia-Keller, C.; Martinez, S.A.; Esparza, M.A.; Bollati, F.; Kalivas, P.W.; Cancela, L.M. Cross-sensitization between cocaine and acute restraint stress is associated with sensitized dopamine but not glutamate release in the nucleus accumbens. Eur. J. Neurosci. 2013, 37, 982–995.
- Caudal, D.; Godsil, B.P.; Mailliet, F.; Bergerot, D.; Jay, T.M. Acute stress induces contrasting changes in AMPA receptor subunit phosphorylation within the prefrontal cortex, amygdala and hippocampus. PLoS ONE 2010, 5, e15282.
- Yuen, E.Y.; Liu, W.; Karatsoreos, I.N.; Feng, J.; McEwen, B.S.; Yan, Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc. Natl. Acad. Sci. USA 2009, 106, 14075–14079.
- Suenaga, T.; Morinobu, S.; Kawano, K.; Sawada, T.; Yamawaki, S. Influence of immobilization stress on the levels of CaMKII and phospho-CaMKII in the rat hippocampus. Int. J. Neuropsychopharmacol. 2004, 7, 299–309.
- Rosa, M.L.; Guimaraes, F.S.; Pearson, R.C.; Del Bel, E.A. Effects of single or repeated restraint stress on GluR1 and GluR2 flip and flop mRNA expression in the hippocampal formation. Brain Res. Bull. 2002, 59, 117–124.
- Elhussiny, M.E.A.; Carini, G.; Mingardi, J.; Tornese, P.; Sala, N.; Bono, F.; Fiorentini, C.; La Via, L.; Popoli, M.; Musazzi, L.; et al. Modulation by chronic stress and ketamine of ionotropic AMPA/NMDA and metabotropic glutamate receptors in the rat hippocampus. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 104, 110033.
- Lin, M.; Hou, G.; Zhao, Y.; Yuan, T.F. Recovery of Chronic Stress-Triggered Changes of Hippocampal Glutamatergic Transmission. Neural Plast. 2018, 2018, 9360203.
- Pillai, A.G.; Arp, M.; Velzing, E.; Lesuis, S.L.; Schmidt, M.V.; Holsboer, F.; Joels, M.; Krugers, H.J. Early life stress determines the effects of glucocorticoids and stress on hippocampal function: Electrophysiological and behavioral evidence respectively. Neuropharmacology 2018, 133, 307–318.
- Hou, X.Y.; Hu, Z.L.; Zhang, D.Z.; Lu, W.; Zhou, J.; Wu, P.F.; Guan, X.L.; Han, Q.Q.; Deng, S.L.; Zhang, H.; et al. Rapid Antidepressant Effect of Hydrogen Sulfide: Evidence for Activation of mTORC1-TrkB-AMPA Receptor Pathways. Antioxid. Redox Signal. 2017, 27, 472–488.
- Xia, B.; Chen, C.; Zhang, H.; Xue, W.; Tang, J.; Tao, W.; Wu, R.; Ren, L.; Wang, W.; Chen, G. Chronic stress prior to pregnancy potentiated long-lasting postpartum depressive-like behavior, regulated by Akt-mTOR signaling in the hippocampus. Sci. Rep. 2016, 6, 35042.
- Kvarta, M.D.; Bradbrook, K.E.; Dantrassy, H.M.; Bailey, A.M.; Thompson, S.M. Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J. Neurophysiol. 2015, 114, 1713–1724.
- Gulemetova, R.; Drolet, G.; Kinkead, R. Neonatal stress augments the hypoxic chemoreflex of adult male rats by increasing AMPA receptor-mediated modulation. Exp. Physiol. 2013, 98, 1312–1324.
- Esparza, M.A.; Bollati, F.; Garcia-Keller, C.; Virgolini, M.B.; Lopez, L.M.; Brusco, A.; Shen, H.W.; Kalivas, P.W.; Cancela, L.M. Stress-induced sensitization to cocaine: Actin cytoskeleton remodeling within mesocorticolimbic nuclei. Eur. J. Neurosci. 2012, 36, 3103–3117.
- Ju, L.; Yang, J.; Zhu, T.; Liu, P.; Yang, J. BDNF-TrkB signaling-mediated upregulation of Narp is involved in the antidepressant-like effects of (2R,6R)-hydroxynorketamine in a chronic restraint stress mouse model. BMC Psychiatry 2022, 22, 182.
- Lumsden, E.W.; Troppoli, T.A.; Myers, S.J.; Zanos, P.; Aracava, Y.; Kehr, J.; Lovett, J.; Kim, S.; Wang, F.H.; Schmidt, S.; et al. Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc. Natl. Acad. Sci. USA 2019, 116, 5160–5169.
- Fukumoto, K.; Fogaca, M.V.; Liu, R.J.; Duman, C.H.; Li, X.Y.; Chaki, S.; Duman, R.S. Medial PFC AMPA receptor and BDNF signaling are required for the rapid and sustained antidepressant-like effects of 5-HT1A receptor stimulation. Neuropsychopharmacology 2020, 45, 1725–1734.
- Li, X.; Witkin, J.M.; Need, A.B.; Skolnick, P. Enhancement of antidepressant potency by a potentiator of AMPA receptors. Cell. Mol. Neurobiol. 2003, 23, 419–430.
This entry is offline, you can click here to edit this entry!