Mitochondria is one of the most prominent sources of reactive oxygen species within a cell which contribute to oxidative stress [
4]. The electron transport chain located on the inner mitochondrial membrane generates the majority of mitochondrial ROS during the process of oxidative phosphorylation (OXPHOS). Leakage of electrons at complex I and complex III from ETC leads to a partial reduction of oxygen to form superoxide which undergoes spontaneous dismutation to hydrogen peroxide, both of which are collectively considered as mitochondrial ROS [
5]. Endogenous modulators such as NO and Ca
2+ have been observed to regulate the production of mtROS by regulating the metabolic states of mitochondria. The mitochondrial Ca
2+ levels increase the rate of electron flow in the ETC and thus decrease mtROS generation [
6]. However mitochondrial Ca
2+ overload increases mtROS production [
7]. STAT3, a transcription factor that regulates gene expression in response to cytokines interleukin (IL)-6 and IL-10, also modulates the activity of the ETC [
8,
9]. Hence a decrease in expression of STAT3 may be correlated to increasing mtROS at complex I [
8]. TNF-α that causes the shedding of TNF-α receptor-1 reducing the severity of microvascular inflammation, has been found to induce a calcium-dependent increase in mt ROS [
10]. Studies have shown that many ROS-producing enzymes, like NADPH oxidase, xanthine oxidase, and uncoupled eNOS, can stimulate mtROS production in a process called “ROS-induced ROS” [
11,
12,
13]. Another transcription factor hypoxia-inducible factor 1α (HIF-1α) also plays a prominent role in bringing about a reduction in ROS by a number of mechanisms including induction of pyruvate dehydrogenase kinase 1 (PKD1), which shunts pyruvate away from the mitochondria; triggering mitochondrial selective autophagy; and induction of microRNA-210 blocking OXPHOS [
14]. Low levels of mtROS regulate the stability of HIF-1α leading to hypoxia adaptation while moderate levels of mtROS have been found to regulate the production of proinflammatory cytokines by directly activating the inflammasome and mitogen-activated protein kinase (MAPK). However, high levels of mtROS are capable of inducing apoptosis by oxidation of the mitochondrial pores and autophagy by the oxidation of autophagy-specific gene 4 (ATG4) [
5]. Depending on the tumor cell microenvironment, the c-Myc gene controls apoptosis by inducing aerobic glycolysis and/or OXPHOS which is required for the activation of certain tumor suppressor proteins, such as Bax and Bak [
15,
16,
17].
Mitochondria also play an important role in the loss of caveolin 1 (cav-1) in the tumor-associated fibroblast compartment, which is related to the early tumor recurrence, metastasis, tamoxifen-resistance, and aggravated increase in tumor growth [
2]. Cav-1 loss induces autophagy and mitophagy, [
18] driving the “Reverse Warburg Effect” by a feed-forward mechanism. This onset of inflammation, autophagy, mitophagy, and aerobic glycolysis in the tumor microenvironment is triggered by activation of the transcription factors NFκB and HIF-1α [
19,
20]. Mitochondria-generated ROS plays an important role in cell proliferation and quiescence. The pro- or anti-tumorigenic signaling is controlled by a mitochondrial ROS switch of the antioxidant SOD2/MnSOD [
21]. Cell proliferation is favored by decreased SOD2/MnSOD activity resulting in increased O
2− production whereas proliferating cells transit into quiescence when SOD/MnSOD activity increases resulting in increased H
2O
2 activity [
22]. Inactivation of mitochondrial antioxidant responses like the Thioredoxin reductase (TrxR); which causes reduction of oxidized Trx to produce reduced Trx that reacts with ROS, contributes to increased oxidative stress in cancer cells.
The increased metabolic requirements of the cancer cells are met by upregulation of glucose transport and metabolism irrespective of oxygen supply [
24]. There is also some evidence that cancer cells decrease mitochondrial respiration in the presence of oxygen, which suppresses apoptosis [
25]. Under hypoxic conditions, the accelerated metabolism produces ROS in cancer cells that is countered by the increased NADPH which is met by the upregulated glycolysis [
26,
27]. NADPH is an essential cofactor for replenishing reduced glutathione (GSH) which is a critical antioxidant. Therefore, not only are cancer cells’ multiple urgent requirements catered to but cancer cell oxidative stress is also controlled by the Warburg effect [
8]. Tumor cells have been reported to switch between the isoforms of pyruvate kinase, used in the last steps of glycolysis [
28]. PKM2 the isoform found in high levels in tumor cells is slower and leads to the accumulation of PEP which in turn activates PPP by feedback inhibition of the glycolytic enzyme triosephosphate isomerase (TPI). This produces more NADPH which reduces ROS and further amplifies the inhibitory effect of PKM2 [
26,
27], Therefore ROS and PKM2 form a negative feedback loop to maintain ROS in a tolerable and functional range. The ROS-regulated gene, hypoxia-inducible factors (HIF-1α) regulates hypoxia-associated genes, some of which are associated with the Warburg effect and its accompanying pathways and hence, are a target of cancer therapies. PKM2 has been found to be the prolyl hydroxylases (PHDs)-induced coactivator for HIF-1α [
8,
29]. HIF-1α also regulates the MYC proto-oncogene which produces MYC protein [
30] that regulates genes participating in energy generation and cell growth and proliferation. HIF-1α and MYC activate hexokinase 2 (HK-II) and pyruvate dehydrogenase kinase 1 (PDK1), which inhibits TCA and increases conversion of glucose to lactate [
31]. Glucose transporter 1 (GLUT1) and lactate dehydrogenase A (LDHA) are also activated by HIF1 and MYC independently, resulting in increased glucose influx and higher glycolytic rates [
13]. Warburg effect increases steady-state ROS condition in cancer cells by producing lactate that is extruded through monocarboxylate transporters to the microenvironment of cancer cells which has no antioxidant properties in contrast to pyruvate, citrate, malate, and oxaloacetate together with the reducing equivalents (NADH.H
+) which are antioxidant intermediates. This increased oxidative stress in cancer cells is stopped from reaching cytotoxic levels by some antioxidant effects exerted by hexokinase II (HK II) and NADPH.H
+ produced through HMP shunt. Latest studies show tumor cells have the capability to carry about both glycolytic and oxidative phosphorylation (OXPHOS) metabolism which makes them resistant to oxidative stress through enhanced antioxidant response and increased detoxification capacity [
32]. The changes related to energy metabolism may be correlated to the expression of certain p53 downstream genes regulated by it, including SCO2, TIGAR, and the p53 inducible gene 3 (PIG3) [
33,
34,
35].
3. Mechanism of Oxidative Stress-Related Carcinogenesis
3.1. Role of ROS in Tumor Cell Proliferation, Survival and Tumor Progression
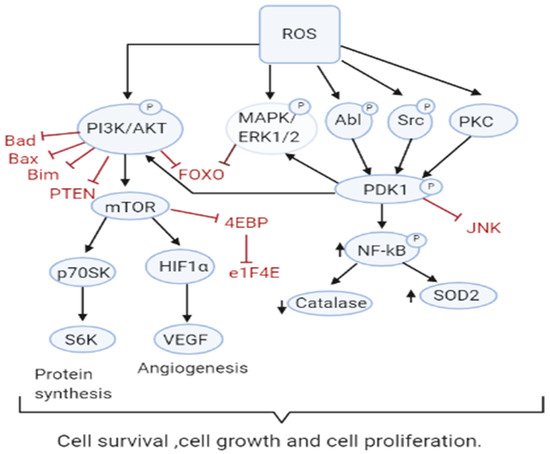
Increased ROS is responsible for the oxidation of negative feedback loop controllers and hence control the actions of other signaling pathways in tumor growth and programmed cell death by the phosphoinositide 3-kinase/protein kinase B (PI3K/PKB) and mitogen-activated protein kinase (MAPK) signaling pathways [
45,
46] (
Figure 2). Reactive oxygen species generation in cancer cells leads to the inactivation of PTEN that leads to an increase in PI3K/Akt signaling that promotes proliferation. Moreover, the cancer cell cycle progression is promoted when ROS inhibits phosphatase Cdc14B resulting in the activation of cyclin-dependent kinase 1 (Cdk1). As mentioned earlier, the major ROS-regulated gene HIF activates PDK1 that further activates Akt which inhibits the tuberous sclerosis complex (TSC). This downregulates mTOR which is a major regulator of cell growth by controlling mRNA translation, ribosome biogenesis, autophagy, and metabolism [
44,
47]. MAPK/ERK1/2 are activated by growth factors and K-Ras stimulated pathways, lead to increased cellular proliferation in cancer cells [
48]. H
2O
2 has also been found to be responsible for the activation of ERK1/2 and pro-survival PI3K/Akt signaling pathway, resulting in increased proliferation [
49,
50]. Studies on breast, leukemia, melanoma, and ovarian cancer have shown ERK1/2 plays additional roles like cell survival, anchorage-independent growth, and motility [
51]. The Akt pathway inactivates pro-apoptotic Bad, Bax, Bim, and Foxo transcription factors by phosphorylation thereby promoting cell survival [
52,
53]. Akt is activated by the Epithelial growth factor (EGF)-derived H
2O
2 production, observed in ovarian cancers [
54]. Cell survival is promoted by the oxidation and inactivation of the negative regulators of PI3K/Akt signaling, i.e., the phosphatases PTEN and PTP1B. The tumor suppressor PTEN has been found to be reversibly inactivated by H
2O
2 in a variety of cancers [
55,
56].
Figure 2. ROS Drive Mitogenic Signaling Cascades. Increased ROS levels contribute to sustained cell survival and proliferation through many pathways including PI3K/AKT, MAPK/ERK1/2, and PKD. ROS also inactivate their downstream targets including Bad, Bax, Bim, Foxo, and PTEN and the JNK pathway.
PKD signaling plays an important role in the detoxification from elevated ROS production and stimulation of anti-apoptotic genes [
57,
58,
59]. PKD1 signaling leads to upregulation of NFκB which also plays an important role in the proliferation and survival of the cell. PKD1 also promotes cell survival through activation of ERK1/2 and down-regulation of the pro-apoptotic c-Jun N terminal protein kinase (JNK) pathway [
60]. An increase in antioxidants SOD2 and Nrf2 has been observed, however, catalase levels appear to decrease providing a role for PKD1 in cell survival [
61]. Other members of the PKD family, PKD2 and PKD3 are implicated to play a role in various other cancers [
60]. The tumor suppressor genes produce proteins that play important roles as antioxidants. For instance, p53 could regulate the expression of various antioxidant enzymes including catalase, SOD2, and GPX1 thereby decreasing ROS accumulation [
62]. However, since p53 is lost or mutated in most cancers, ROS accumulation and pro-tumorigenic signaling is found.
3.2. Role of ROS in Apoptosis-Tumor Suppressive Role
Though ROS activates mitogenic signaling pathways, high levels of ROS have the ability to induce cell cycle arrest, senescence, and cancer cell death either by the initiation of intrinsic apoptotic signaling in the mitochondria or by extrinsic apoptotic signaling by the death receptor pathways [
63]. ROS induces apoptosis by activating ASK1/JNK and ASK1/p38 signaling pathways in human cancer cells [
64,
65]. These pathways are activated when TRX1 is oxidized by H
2O
2 which subsequently dissociates from ASK1, thereby activating the downstream MAP kinase kinase (MKK)4/MKK7/JNK and MKK3/MKK6/p38 pathways leading to suppression of anti-apoptotic factors [
17,
66,
67,
68]. It has also been shown that ROS mediate the downregulation of FLICE inhibitory proteins (FLIP proteins) by ubiquitination and subsequent degradation by the proteasome and thereby induce apoptosis by Fas ligand activation [
69]. Collectively, these observations support a tumor-suppressive role of ROS [
70]. Recent studies have shown that p53 plays an important role in oxidative stress-related cell death. A regulatory signaling protein of phosphatidyl-3-OH kinase (PI (3) K), p85, participates in the cell death induced by oxidative stress independent of PI (3) K [
71]. This protein p85 is upregulated by p53. Sir2α has been found to interact with p53 and attenuate p53-mediated functions and hence is a potential cancer therapeutic target [
72].
JNK pathway activation by elevated ROS production results in apoptosis initiated by intrinsic apoptotic signaling through mitochondria or extrinsic apoptotic signaling mediated by death receptor pathways [
73,
74,
75]. JNK pathway mutations have been found to be inactivated in various cancers suggesting that these pathways may be implicated in apoptotic signaling [
76]. The activity of apoptotic effectors including the Bcl-2 family of proteins and cytochrome c are affected by the overproduction of ROS leading to the activation of the caspases, a prominent hallmark of apoptosis, resulting in the cleavage of poly ADP ribose polymerase (PARP), DNA fragmentation, and cell death [
77].
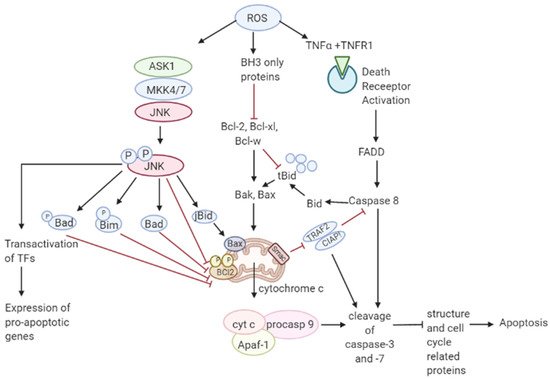
As mentioned in
Figure 3, elevated ROS can also result in apoptosis by binding of ligands to death receptors which trigger caspase activation of the initiator caspase 8 leading to cleavage of downstream caspase 3 and Bcl-2 protein Bid to tBid which then translocate into the mitochondria causing the release and translocation of cytochrome c [
78,
79]. Cytochrome c forms a complex with apoptotic protein-activating factor 1 (Apaf-1) and pro-caspase 9 inducing the cleavage of downstream caspase-3 and -7. Members of the Bcl-2 family, anti-apoptotic (Bcl-2, Bcl-w, and Bcl-xL) are inhibited, and pro-apoptotic (Bad, Bak, Bax, Bid, and Bim) are activated in apoptotic signaling [
80]. The loss of cytochrome c from the mitochondria will disrupt and damage the mitochondrial ETC and further cause elevated production of ROS [
81]. ROS-induced apoptosis can be attributed mainly to decreased GSH levels and the loss of redox homeostasis [
82].
Figure 3. Role of ROS in apoptosis. Toxic ROS levels damage the mitochondrial membrane releasing cytochrome c to the cytoplasm which forms a complex with Apaf-1 and pro-caspase 9. This induces the cleavage of caspase-3 and -7 resulting in apoptosis. Additionally, binding of TNFα ligand to TNFR1 death receptor triggers the activation of caspase 8 leading to cleavage of caspase 3. Caspase 8 activation also cleaves Bcl-1 protein Bid to form tBid which further leads to the release of cytochrome c in the intrinsic apoptotic pathway.
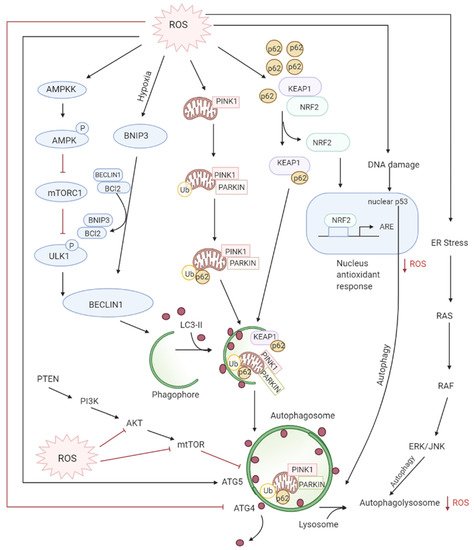
3.3. Role of ROS in Autophagy-Both Tumor Suppressive and Tumor Promoting Roles
Autophagy is the controlled lysosomal pathway that regulates cellular homeostasis by degradation and recycling of proteins and organelles within a cell [
83]. ROS regulates autophagy in both direct and indirect ways. Direct regulation involves modification of key proteins like Atg4, Atg5, and Beclin which are involved in the autophagy process. Indirect regulation by ROS involves alteration of signaling pathways that can induce autophagy such as the JNK, p38. ROS have also been found to inhibit Akt signaling and downstream mTOR and thereby induce autophagy [
84]. Autophagy is one of the first defenses against oxidative stress damage and is upregulated in response to elevated ROS levels [
85]. Autophagy has been found to be regulated by the mammalian target of rapamycin complex1 (mTORC1) and its upstream activators PI3K and AKT that suppress autophagy whereas negative regulator of PI3K and AKT pathways PTEN has been found to induce autophagy [
86]. DNA damage caused by the ROS produced by mitochondria leads to activation of p53 that has been documented to regulate autophagy [
87] (
Figure 4).
Figure 4. ROS levels regulate autophagy levels by different pathways: firstly, oxidation of ATG4 leads to accumulation of autophagosomes, secondly, the AMPK signaling cascade induces autophagy through the ULK1 complex. Thirdly, the disruption of BCl-2-BECLIN interactions also initiates autophagy. Lastly, the alteration of mitochondria homeostasis leads to mitophagy activation which checks ROS accumulation by elimination of damaged mitochondria. The degradation of KEAP1 by selective autophagy mediated by p62 leads to the expression of Nrf2-regulated antioxidant genes thereby reducing ROS.
Deregulated autophagy has been found to have a role both in tumor progression and tumor suppression. During the early steps of cancer development, autophagy inhibits tumorigenesis by preventing ROS-induced damages on DNA and protein. However, during the later stages of cancer development (promotion, progression, and metastasis), autophagy plays a pro-tumoral role by eliminating ROS-induced metabolic stress and producing nutrients required for cancer cell survival. The cancer cells under hypoxia induce the formation of ROS which can activate autophagy in neighboring stroma cells which then provide high-energic nutrients, such as lactate or ketones, necessary for cancer cell survival and proliferation in accordance with what we have seen earlier, also termed as “tumor-stromal co-evolution”.
Increased ROS production induces autophagy of damaged mitochondria called mitophagy restoring the physiological ROS levels. [
88] This selective autophagy is mediated by two different molecular pathways: NIX/BNIP3L and PARKIN (PARK2)/PTEN induced putative kinase 1 (PINK1) [
89,
90,
91,
92]. Nix/BNIP3L targets mitochondria for degradation after interacting with GABARAP and GABARAPL1 at the autophagosome [
93,
94]. Whereas selective degradation of damaged dysfunctional mitochondria occurs through PARKIN/PINK1 after ROS induces mitochondrial membrane depolarization [
92]. Moreover, Nrf2/Keap1 and SQSTMI/p62 pathways also regulate mitophagy by decreasing ROS [
95]. SQSTM1/p62 interacts with Nrf2/Keap1, forming a complex with Keap1, preventing Nrf2 degradation resulting in the release and translocation of Nrf2 to the nucleus where it activates antioxidants [
96,
97].
Studies have proven that elevated ROS levels can also lead to defective autophagy. Deletion of autophagy genes ATG5 or ATG7 leads to autophagy inhibition and accumulation of damaged mitochondria which results in chronic oxidative stress, tissue damage, and inflammation which all favor tumor initiation [
97,
98,
99]. BECLIN1 (ATG6/BECN1) which is an essential gene in autophagy as well as a tumor suppressor has been found deleted in various cancers, resulting in damage to mitochondria, oxidative stress, and disease progression [
100,
101,
102]. Furthermore, during later stages of tumor initiation, autophagy is required for cell transformation by the RAS oncogene in order to promote cell tolerance to stress, therefore for Ras-induced tumorigenesis, active autophagy is necessary to maintain cellular homeostasis [
83].