Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Ischemic stroke is a devastating disease induced by partial or total occlusion of a cerebral artery, the middle cerebral artery being that most frequently affected in humans. Stroke is now the leading cause of disability and the second cause of death worldwide. Ischemia accounts for 80% of all strokes.
- mTOR
- brain ischemia
- neuron
1. Introduction
Mammalian/mechanistic target of rapamycin (mTOR) is a 289 kDa serine–threonine kinase and a key element of two mTOR complexes called mTORC1 and mTORC2 (mTORCs) [1,2,3,4]. Furthermore, mTOR is highly conserved and is the center of multiples signaling pathways and coordinates important cellular processes such as cell growth and metabolism [5]. Although mTOR is ubiquitously expressed, it is especially abundant in the brain [6]. Therefore, mTOR dysfunction profoundly affects the central nervous system (CNS). Mutations in genes encoding mTOR regulators induce neurological disorders called “mTORopathies” [5].
Furthermore, mTOR, as indicated by its name, is a target protein of rapamycin, an immunosuppressant and anti-fungal macrolide compound isolated from Streptomyces hygroscopicus. This kinase comprises several functional domains, including C-terminal small FAT domain (FATC), C-terminal kinase domain (KD), FKBP12 rapamycin-binding domain (FRB), transactivation/transformation-associated domain (FAT), and an N-terminal domain containing at least 20 HEAT (Huntingtin elongation factor 3 A subunit of PP2A TOR1) repeats. The latter provide sites for the interaction of regulatory proteins to form mTORC1 and mTORC2. The KD domain of mTOR, with conserved sequences homologous to the catalytic domain of the phosphoinositide 3-kinase (PI3K) family, contains phosphorylation sites that regulate the activity of this kinase [7].
Rapamycin and its analogs (called rapalogs) act as allosteric inhibitors of mTORC1 by interacting with the FRB domain of mTOR via FKBP12 protein (FK506-binding protein 1 A 12 kDa) [6]. Furthermore, mTORC2 is insensitive to rapamycin inhibition, as initially described [8], but prolonged exposure to this macrolide results in the disruption of the assembly and integrity of mTORC2, thereby causing the functional inhibition of the complex [9].
Additionally, mTORC1 and mTORC2 share several common proteins, including the catalytic subunit mTOR, Deptor (DEP-domain-containing mTOR interacting protein), mLST8 (mammalian lethal with Sec13 protein 8), and Tti1/Tel2 complex [10] (Figure 1). In addition, each complex has specific proteins. Raptor (regulatory-associated protein of mTOR) and PRAS40 (proline-rich Akt substrate 40 kDa) are specific subunits of mTORC1, while Rictor (rapamycin-insensitive companion of mTOR), mSin1, and Protor1/2 are exclusive to mTORC2 [7] (Figure 1). All of these proteins have different functions in the complexes. Not only do they have structural functions (stabilizing the complexes and recruiting mTOR substrates) but they also contribute to regulating mTOR activity. Recently, an advancement in our understanding of the precise functions of each mTOR companion protein beyond the strictly structural function has occurred. Recent studies demonstrate an important impact in the fine-tuned activity of mTOR kinase, according to post-translational modifications of some of the companion proteins, mainly by phosphorylation.
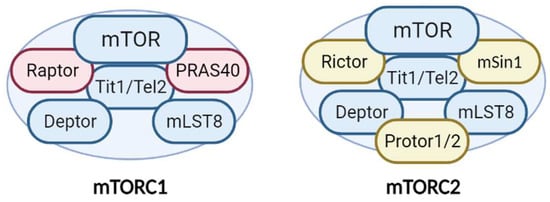
Figure 1. Structure and components of the mTORC1 and mTORC2 complexes. The mTORCs share common proteins (blue) called mTOR (catalytic subunit), Deptor (mTOR inhibitor subunit), mLST8 (scaffold and activator subunit), and Tit/Tel2 (assembly subunit). The specific mTORC1 proteins (pink) include Raptor and PRAS40 (mTOR inhibitor). Rictor, mSIN1, and Protor1/2 (activity modulator) comprise mTORC2 (yellow). See text for more details.
2. mTOR after Cerebral Ischemia
Ischemic stroke is a devastating disease induced by partial or total occlusion of a cerebral artery, the middle cerebral artery being that most frequently affected in humans [93]. Stroke is now the leading cause of disability and the second cause of death worldwide [93,94]. Ischemia accounts for 80% of all strokes.
The occlusion of a cerebral artery dramatically reduces the blood flow to several brain regions (Figure 5). As a result, there is a decrease in the supply of nutrients (glucose), oxygen, and growth factors to neurons. These conditions lead to neuronal injury or death, bringing about serious neurological dysfunction, the severity of which depends on the size of the area affected in the brain and the duration of the occlusion [95].
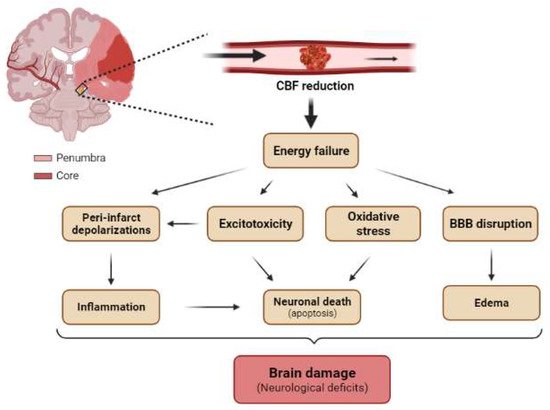
Figure 5. Main pathological mechanisms of brain ischemia at tissue levels. Schematic representation of a coronal section of the adult human brain with occlusion of MCA showing the core (red) and the penumbra (light red) regions. The reduction in cerebral blood flow (CBF) induces energetic failure at the tissue level, which promotes excitotoxicity, peri-infarct depolarization, oxidative stress, and BBB disruption. Secondarily, these pathological mechanisms triggers neuroinflammation, neuronal death (mainly by apoptosis), and cerebral edema, which in turn trigger tissue damage and neurological deficits.
To date, the only therapeutical intervention available to ameliorate ischemic damage is a reduction in neuronal injury via reperfusion using a thrombolytic agent (recombinant tissue plasminogen activator, tPA) or surgical removal of clots [84]. However, these strategies are very limited because they are only feasible within a short time window after ischemia onset due to the high risk of intracerebral hemorrhage, which would worsen the prognosis of the patient. Given these considerations, reperfusion is suitable for only a few patients (approximately 5%) [96]. Although reperfusion can effectively decrease the infarct volume and improve neurological recovery, it does not reduce the subsequent neurodegeneration that occurs after brain ischemia. Moreover, ischemia–reperfusion injury occurs, consisting of multiple pathological events, such as excitotoxicity, oxidative stress, inflammation, apoptosis, and blood–brain barrier (BBB) disruption [97]. In this regard, it is necessary to develop new therapeutic strategies to alleviate ischemic damage in situations with or without reperfusion.
Most of the neuroprotective compounds that have been effective in animal models are ineffective in humans. This has led to doubts about the suitability of in vivo models of stroke. However, we consider that the differences in the responses of animal models and humans to these agents reflect the complex mechanisms that underlie cerebral ischemia and highlight the need for a greater knowledge of the same. Along these lines, a deeper understanding of the molecular mechanisms triggered after cerebral ischemia will provide better opportunities to discover new therapeutic targets. In this regard, mTOR emerges as a potential candidate.
Two distinct approaches are used to study the effects of ischemia on the brain. The most common is an in vitro model, which involves primary cultures of neural cells exposed to oxygen and glucose deprivation (OGD). According to the literature available, the duration of OGD fluctuates from 30 min to 6–12 h. However, the percentage of oxygen remains more constant between the different studies, with 1% being the most common value used. Differences in the exposure to OGD could explain the observed discrepancies with respect to the role of mTOR in this model of ischemia. This in vitro model allows the study of the individual cellular and molecular changes that take place in each type of brain cell after ischemia. Nevertheless, the model cannot be used to analyze the relationship between cell types.
The second type of approach used to examine the effects of ischemia on the brain involves in vivo model. They can be divided into two main groups on the basis of the extent of the affected tissue. The first group is global ischemia (two- or four-vessel occlusion), which reproduces the impact of a cardiac arrest on the brain and affects the entire brain. The second group, called focal cerebral ischemia, mimics occlusion of a cerebral artery either by an embolus or local thrombosis, with middle cerebral artery occlusion (MCAo) being the most frequent [84,98,99]. MCAO models can be subdivided into two groups: transient MCAo (tMCAo) and permanent MCAo (pMCAo). The tMCAo is when the occlusion of the artery is sustained for a short period (between 30–120 min) and reperfusion is allowed after. In pMCAo models, the occlusion of MCA is maintained until animal sacrifice. The tMCAo mimics a therapeutic intervention (reperfusion) in stroke patients, while pMCAo does not reproduce a therapeutic intervention.
Ischemic brain injury is the result of a complex sequence of pathophysiological events that progress over time called the “ischemic cascade”. The main pathogenic mechanisms that evolve include excitotoxicity, peri-infarct depolarization, inflammation, and programmed cell death (apoptosis) (Figure 5) [99]. The affected region is not damaged homogeneously after focal ischemia. The area severely affected by hypoperfusion, named the ischemic core, presents a high rate of neuronal death by necrosis (Figure 5). The less affected region surrounding the core is called the penumbra and is characterized by apoptosis, which can be rescued (Figure 5) [95]. Given these considerations, one of the most important targets in neuro-regenerative approaches after ischemia is the penumbra region. The duration of occlusion determines the severity of damage and the prognosis of the patients [100]. The number of surviving neurons after a stroke defines cerebral functional deficits. In this regard, protecting neurons from death has been the focus for recovering cerebral functions after ischemia. Several approaches to achieve this goal include strategies to prevent neuronal death (neuroprotection) and others directed at neuronal repair; that is, neuron-based approaches. The current understanding of glial cell responses after cerebral ischemia reveals that these cells offer an interesting alternative strategy to protect or recover neurons and brain function.
At the tissue level, mTORC2 activity, measured by phosphorylation levels of its main substrate, pAkt-Ser473 (Figure 4), decreases in response to ischemia. Sustained mTORC2 inhibition in pMCAo models has been described, even days after the insult [101,102]. A decrease in mTORC1 activity, measured as a reduction in phosphorylation levels of its major targets P70S6K and 4EBPs (see Section 1.3 and Figure 4), has also been observed in pMCAo models [103,104]. Several studies have revealed a positive neuroprotective effect of mTORCs upregulation after cerebral ischemia [105,106]. In the tMCAo model, mTORC1 and mTORC2 activity suppression using Rapalink-1, a third-generation mTOR inhibitor, has been reported to exacerbate neuronal damage induced by ischemia in the short-term, worsen BBB stability, and increase the area of damage [107]. These observations suggest that the upregulation of mTORC activity after cerebral ischemia has beneficial effects on the CNS.
Many studies on mTOR and cerebral ischemia have focused on autophagy, which plays a critical role in the maintenance of proteostasis and the survival of neurons by promoting the lysosome-driven removal of damaged or non-essential molecules and defective organelles (see Section 1.3). Canonically, autophagy is activated by starvation conditions, and consequently it induces the elimination of organelles and molecules to compensate. Autophagy is a key mechanism to protect neurons against ischemia. Treatment with rapamycin after MCAo decreases mTORC1 activity and increases autophagy, thereby reducing neuronal apoptosis [83,104]. In animal models, preconditioning with rapamycin improves brain tolerance to ischemic damage [108], ameliorates neurological deficits, and reduces infarct volume and brain edema [109,110,111]. Rapamycin administration before or after tMCAo reduces infarct volume and neurological dysfunction [83]. However, the role of autophagy in cerebral ischemia remains controversial, since current lines of evidence suggest that overactivation of autophagy induces cell death and aggravates ischemic brain injury [73,85]. Several data from in vivo models support the notion that autophagy suppression through the AMPK/mTORC1 pathway after ischemia–reperfusion (tMCAo models) reduces cerebral damage and brain edema, improving the neurological score [112,113,114]. This overactivation of autophagy is probably triggered by a dramatic decrease in mTORC1 activity through the upregulation of AMPK and downregulation of the PI3K/Akt pathway induced by the ischemic conditions. Post-ischemic treatment using specific autophagy inhibitors significantly decreases the volume of the injured area in global ischemia [115]. Furthermore, the reduction in autophagy through mTORC1 activation has proven beneficial in tMCAo models, in which reperfusion is allowed and the availability of nutrients and oxygen is restored. It would be interesting to analyze the role of autophagy in pMCAo models, in which the reestablishment of normal conditions does not occur.
The effects of cerebral ischemia are far from homogeneous. It should be noted that cerebral ischemia triggers the activation of the mTORC1 pathway in the ischemic penumbra and a decrease in the ischemic core [83]. This is an important consideration to understand the effects of rapamycin treatment on in vivo models of ischemia, as the ischemic core and penumbra regions change over time.
3. mTORCs and Neurons
Neurons are the neural cells that are most sensitive to ischemic damage. Most studies addressing cerebral ischemia have focused on attempts to reduce neuronal death to ameliorate damage. Unfortunately, the promising results obtained in animal models using neuroprotective agents have not been reproduced in humans [95].
The role of mTORC1 and mTORC2 in neurons under ischemic conditions has been widely studied [20,117,118]. As we described previously, mTORC1 activity is finely tuned by multiple upstream signaling pathways (see Section 1.2). Ischemia dramatically reduces mTORC1 activity in neurons through the dysregulation of all of its upstream pathways (Figure 3) [34,103,119,120]. Many studies suggest that the canonical PI3K/Akt pathway is the most influential with respect to reducing neuronal mTORC1 activity after ischemia [121,122]. Ischemia promotes the depletion of growth and extracellular survival factors that downregulate the PI3K/Akt pathway, thereby inhibiting mTORC1 and triggering a reduction in phosphorylation levels of neuronal P70S6K, affecting protein synthesis and neuronal viability (Figure 6) [123,124,125]. Thus, recovery of mTORC1 activity through the activation of the neuronal survival PI3K/Akt pathway reduces the loss of neurons [101,102,103,126,127,128,129,130]. Additionally, the exposure of primary neurons to AZD2014, an mTOR inhibitor, after OGD induces an increase in neuronal death via the downregulation of the TSC/mTORC1 pathway [131].
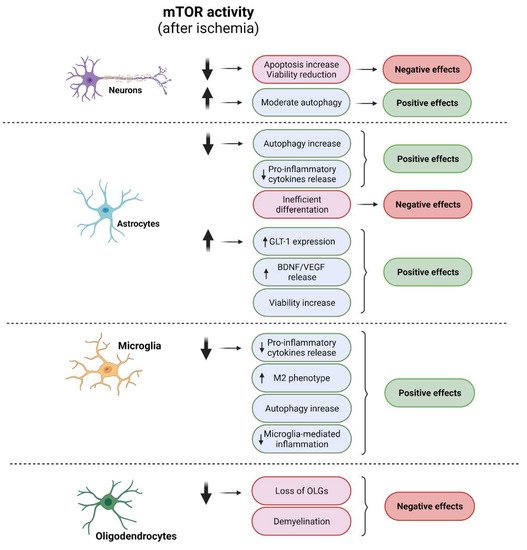
Figure 6. Cellular effects of modulation of mTOR in ischemic conditions. Diagram of the impacts of mTOR modulation in each neural cell type and their correlation with specific cellular processes. Positive effects are shown in green and negative effects in red. A diminution of mTOR activity after ischemia induces negative effects in neurons and OLG mainly related with a viability reduction, whereas an increment of this kinase activity triggers positive effects. In microglia, a reduction in mTOR activity shows positive effects via the induction of M2 phenotype and reduction in microglia-mediated inflammation. In astrocytes, either an activation of or reduction in mTOR activity shows positive effects.
Rapamycin is the most commonly used inhibitor of mTOR and a known autophagy inductor. An increase in autophagy or autophagic flux by rapamycin reduces neuronal apoptosis after OGD or reperfusion injury [119,132]. Zhang et al. [132] proposed a complex relationship between neuronal apoptosis and autophagy that could explain the beneficial effects observed with rapamycin.
These seemingly contradictory results regarding the positive effects of either activation or inhibition of mTOR in neurons in the context of ischemia may be explained by the doses and time points selected for the analysis. Moreover, the signaling pathways chosen to impact mTOR activity could represent a source of high variability in the outcome. Related to this, upregulation of the survival pathway PI3K/Akt is known to have beneficial effects not only through mTORC1 activation, but also by modulating other relevant targets described to improve neurons survival, such as CREB, FOXO, or GSK3, among others [133,134]. On the other hand, a direct inhibition of mTOR by rapamycin both pre- and post-OGD improves neuronal viability via induction of autophagy. However, only low doses of rapamycin have been shown to be beneficial, while high doses prove toxic, probably by affecting mTORC2 activity and reducing neuronal survival [135].
Little information is available regarding mTORC2 activity in neurons. This complex is downregulated after brain ischemia, measured as a reduction in the phosphorylation levels of Akt-Ser473 [20,103,136], while increases in its activity have beneficial effects after damage [137].
4. mTORCs and Glial Cells
Glial cells are abundant in the mammalian CNS. However, their role in physiological and pathological conditions is not yet fully understood. As we gain a greater understanding of glial cells, their relevance increases. In this context, these cells emerge as potential therapeutic targets. However, limited data are available to support a relationship between glial cells and the activity of mTORC1 and mTORC2.
4.1. Astrocytes
Astrocytes are the most abundant cell type in the CNS, accounting for 50% of the human brain volume [138]. There are two main groups that differ in morphological and spatial distribution: fibrous astrocytes, which are predominant in white matter; and protoplasmic astrocytes, predominant in gray matter [139]. The functional role of each type after ischemic injury is unknown, so the term “astrocyte” is used herein to include both types. Astrocytes play an essential role in multiple physiological and pathological processes of the CNS, beyond the initial notion that they are mere supportive cells to maintain functional neurons and neuronal circuits.
After cerebral ischemia, astrocytes have been associated with several positive effects, such as BBB stability, neuronal metabolic support, and neuroprotection. One well-known mechanism of astrocyte-driven neuroprotection is the capacity of these cells to reduce excitotoxicity by decreasing excessive extracellular glutamate released by damaged neurons. This decrease is achieved through the activation and overexpression of various astrocytic glutamate transporters, such as glutamate transporter-1 (GLT-1) [95]. In addition, astrocytes are involved in the management of oxidative stress [140].
The release of distinct inflammatory factors by ischemic neurons and reactive microglia induces astrocytes to switch to a reactive status, which can have a negative or positive effect on ischemic damage depending on the phenotype they acquire. Like microglia, astrocytes can acquire an A1 (pro-inflammatory) or A2 (anti-inflammatory or neuroprotector) phenotype. Reactive astrocytes reduce the detrimental accumulation of reactive oxygen species (ROS) during ischemia, thereby reflecting their potential role as anti-oxidative players under pathological conditions [95,141,142]. While there is extensive information about the role of astrocytes after cerebral ischemia, few studies have described the participation of mTOR activity in this context. Data currently available on the positive or negative role of mTORCs in astrocytes after cerebral ischemia are contradictory.
Some studies show the beneficial effects of mTOR activation on astrocytes after OGD (Figure 6). This upregulation of mTORC1/S6K1 contributes to astrocyte survival after ischemia [125]. In addition, an increase in astrocytic mTOR activity after OGD through the activation of the PI3K/Akt pathway triggers the astrocytic release of VEGF and BDNF, which promote angiogenesis and enhance neuron survival [143]. Furthermore, mTOR activation has been reported to increase GLT-1, which promotes glutamate uptake and reduces the excitotoxicity of neurons [144,145,146]. All of the above-mentioned data suggest that mTOR activation in astrocytes has a positive effect within the context of ischemic damage.
However, a beneficial effect of astrocytic mTOR inhibition under ischemic conditions has been demonstrated (Figure 6). The reduction in astrocytic mTORC1 activity via AMPK [147,148,149] or TSC2 [150] after OGD induces an increase in autophagic flow and a decrease in the release of pro-inflammatory cytokines [151,152], thereby improving neuronal viability.
There are limited data regarding the role of astrocytic mTORC2 after ischemia. The increase in ROS after acute prenatal hypoxia triggers the inhibition of mTORC2 activity by ubiquitination and degradation of Rictor. This inhibition then leads to the inefficient differentiation of astrocytes, thereby affecting their response to ischemia and worsening damage to the brain [153]. After OGD, full mTORC2 activity is required for the overexpression of GLT-1, which is beneficial after ischemia by reducing excitotoxicity [145].
4.2. Microglia
Microglia are the first line of defense against cerebral damage and infection by pathogens. These cells are uniformly distributed throughout the brain [4] and they account for 10–15% of all cells in this organ. Under physiological conditions, microglia are in a “resting state”, using their highly motile processes to check the microenvironment of nervous tissue to generate an appropriate response and maintain tissue homeostasis [154]. After cerebral ischemia, this homeostasis is disrupted and microglia respond.
After 2 days of pMCAo, the inflammatory stage is established. Ischemia-induced cell death results in the release of damage-associated molecular patterns (DAMPs), thereby activating microglia to acquire a reactive phenotype [84,101,136,155]. Given the phagocytic capacity of microglia as scavengers, they are the first glial cells to respond to cerebral ischemia [156]. The activation of microglia involves three processes, namely morphological transformation, migration to the damaged area, and proliferation [136].
Two phenotypes of active microglia, namely M1 and M2, have been identified. The M1 phenotype presents a pro-inflammatory profile and can release inflammatory cytokines such as IL-1 and TNFα. As a general concept, this phenotype exacerbates the inflammatory response and ischemic damage [95]. Accordingly, the inhibition of microglial activation by minocycline has been shown to protect the brain against focal ischemia by reducing the expression of IL-1β-converting enzyme and cyclooxygenase-2 [157]. M2 microglia release anti-inflammatory cytokines such as IL-10 and TGF-β, thereby promoting BBB stability and allowing functional recovery of the brain after ischemia [158,159]. An imbalance towards the M1 phenotype is related to poorer clinical prognosis [160]. In this context, most therapies based on microglia are focused on polarizing them towards the M2 phenotype to minimize the deleterious effects of the M1 phenotype [95,161,162]. Despite this classification, many studies support the notion of great heterogeneity in the population of activated microglia and the coexistence of intermediate phenotypes. This complexity hinders advances in our understanding of microglia after damage [163].
The mTORC1 pathway has recently been reported to be involved in microglial activation and their polarization towards the M1/M2 phenotype after ischemia. Furthermore, mTORC1 is a known regulator of immune responses. Some studies addressing mTORC1 inhibition after ischemia reveal promising results, including the reduction in the ischemic area and the amelioration of neurological deficits in animals. The direct inhibition of mTORC1, using pharmacological (sirolimus and everolimus treatment for 6 h after tMCAo induction) and genetic (Raptor-KO mice) approaches, leads to a reduction in pro-inflammatory cytokine expression, in parallel to an increase in the M2 phenotype [164]. Indeed, recent studies show similar results using different approaches to reduce mTORC1 activity, including the downregulation of some upstream players of the PI3K/mTORC1 pathway [165,166,167,168]. The mTOR activity regulates the synthesis of pro-inflammatory cytokines (Figure 6) through the mTOR/STAT3 pathway but also enhances autophagy in microglia [166,169]. Using tMCAo rats, it has been shown that early downregulation of the Akt/mTOR/STAT3 pathway after ischemia suppresses microglia-mediated neuroinflammation and improves cerebral injury [165].
4.3. Oligodendrocytes
Oligodendrocytes (OLGs) are responsible for axonal myelination in the CNS. Myelination, which occurs during embryonic development and continues into adulthood, determines the speed of nerve impulse conduction and supports axons [170]. Myelination is a complex and organized process with high metabolic demands [171]. In this regard, mTORC1, a pivotal player in the coordination of cell metabolism, is involved in this process. Some studies using both in vitro and in vivo models have demonstrated that mTORCs are fundamental players in myelination and OLG differentiation [3,172,173]. Several in vivo approaches using loss-of-function transgenic mice have unveiled the role of mTORC1 and mTORC2 in myelination. Knockout mice for Raptor or Rheb1, in which mTORC1 activity is abolished, show hypomyelination [3,71,172,174]. The same phenotype was observed in Rictor knockout mice, which show suppressed mTORC2 activity [3]. Ablation of mTOR, which affects the function of both complexes, also induces hypomyelination in the CNS [175]. This observed hypomyelination effect does not occur equally in all brain regions, with the cerebellum and spinal cord being more sensitive to mTORC ablation than other regions [176]. We conclude that the combined activity of mTORC1 and mTORC2 is necessary to maintain adequate myelination levels. Of note, the loss of mTORC1 function has a greater impact on myelination than the ablation of mTORC2, and simultaneous inactivation of the two complexes has a summative effect compared to the disruption of mTORC1 alone [172].
Like other cell types, in OLGs, mTORC1 regulates protein and lipid synthesis, both necessary processes for correct myelin synthesis. The inhibition of mTORC1 induces a reduction in the synthesis of myelin proteins by OLGs, thereby impairing myelination [3]. Conversely, depending on the upstream pathway affected, mTORC1 activation in OLGs induces a hyper- or hypo-myelinated phenotype. The overstimulation of Akt in OLGs triggers mTORC1 activation and enhances myelination, with no changes in the proliferation or survival of oligodendrocyte precursor cells (OPCs) or mature OLGs [177,178]. However, the overactivation of mTORC1 through deletion of the inhibitory complex TSC causes hypomyelination, which can be reversed by the administration of rapamycin [172,179]. In line with these results, the brains of patients with tuberous sclerosis (a disorder caused by loss of function mutations of TSC) show impaired white matter integrity [180]. The mechanism underlying this hypomyelination is not understood. It has been reported that mTORC1 overactivation by TSC ablation triggers a reduction in mTORC2 activity, as indicated by lower mTORC2-dependent phosphorylation levels of p-Akt-Ser473 [176,181]. Furthermore, mTORC2 has been associated with lipid metabolism [63] and specifically with the biosynthesis of sphingolipids, an abundant constituent of myelin [182]. This observation could explain the stronger defects in myelination observed in the aforementioned OLG double-Raptor−/− and -Rictor−/− mutants [172].
In addition to their participation in myelination, mTORCs have significant involvement in the differentiation and maturation of OLGs. Using in vitro models, it has been demonstrated that an increase in mTORC1 activity induces the differentiation of OLGs from OPCs. Conversely, the inhibition of mTORC1 using rapamycin prevents OLG differentiation [173,183]. In vivo models reinforce these findings. Conditional ablation of Rictor or Raptor has a differential impact on OLG differentiation. Furthermore, mTORC1 is a positive regulator of OLG maturation through an unknown mechanism, since ablation of Raptor induces an increase in OPC numbers, a selective decrease in myelin protein, and a reduction in the number of mature OLGs in the corpus callosum [3,176]. In contrast, Rictor ablation has a modest effect on OLG differentiation [172].
Cerebral ischemia induces a dual response in the OLG population. During the acute phase, a loss of OLGs occurs in the damaged area, since these cells are highly sensitive to oxidative stress, which occurs in this early stage of injury [184]. Some authors correlate the loss of OLGs and demyelination with a reduction in PI3K/Akt/mTORC1 activity (Figure 6), since the activation of this pathway via downregulation of PTEN enhances myelination and improves neuro-functional recovery from stroke [185]. In the long term, ischemia induces an increase in the number of OLGs, mainly in the penumbra [186,187]. This effect has been related to the activity of the PI3K/Akt/mTOR pathway [188]. Thus, an increase in the activity of this pathway after ischemia reduces the loss of OLGs and myelin, thereby improving neurological deficits (Figure 6) [185,189,190].
The “oligovascular unit”, which defines a dynamic structural complex composed of OPCs and endothelial cells (ECs), is gaining relevance. Interactions between OPCs and ECs play a pivotal role in angiogenesis in both physiological and pathological conditions [191]. After ischemia, the close communication between ECs and OPCs could be directed to recover myelin in the damaged area. In vivo treatment using EC secretomes induces beneficial effects on white matter, such as enhanced vascularization, induction of myelinization, and increases in the number of mature OLGs, thereby improving cognitive function [192]. The ischemia-induced proliferation of OPCs in the oligovascular unit is mediated by the PI3K/Akt/mTOR pathway [193].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23052814
This entry is offline, you can click here to edit this entry!