Chromosomes are organized in distinct nuclear areas designated as chromosome territories (CT). The structural formation of CT is a consequence of chromatin packaging and organization that ultimately affects cell function. Chromosome positioning can identify structural signatures of genomic organization, especially for diseases where changes in gene expression contribute to a given phenotype. The term “chromosome territories” was first coined by Theodor Boveri (1909) in the 20th century. However, the idea of a territorial-like organization of chromosomes during interphase appeared as early as 1885, described by Carl Rabl, based on his experiments of cell division using Salamandra maculata. Rabl observed a polarized nuclear position of chromosomes at the beginning and at the end of mitosis, suggesting a preserved chromosome position during cell cycle phases.
- multiple myeloma
- nuclear organization
- genome markers
- chromosome territories
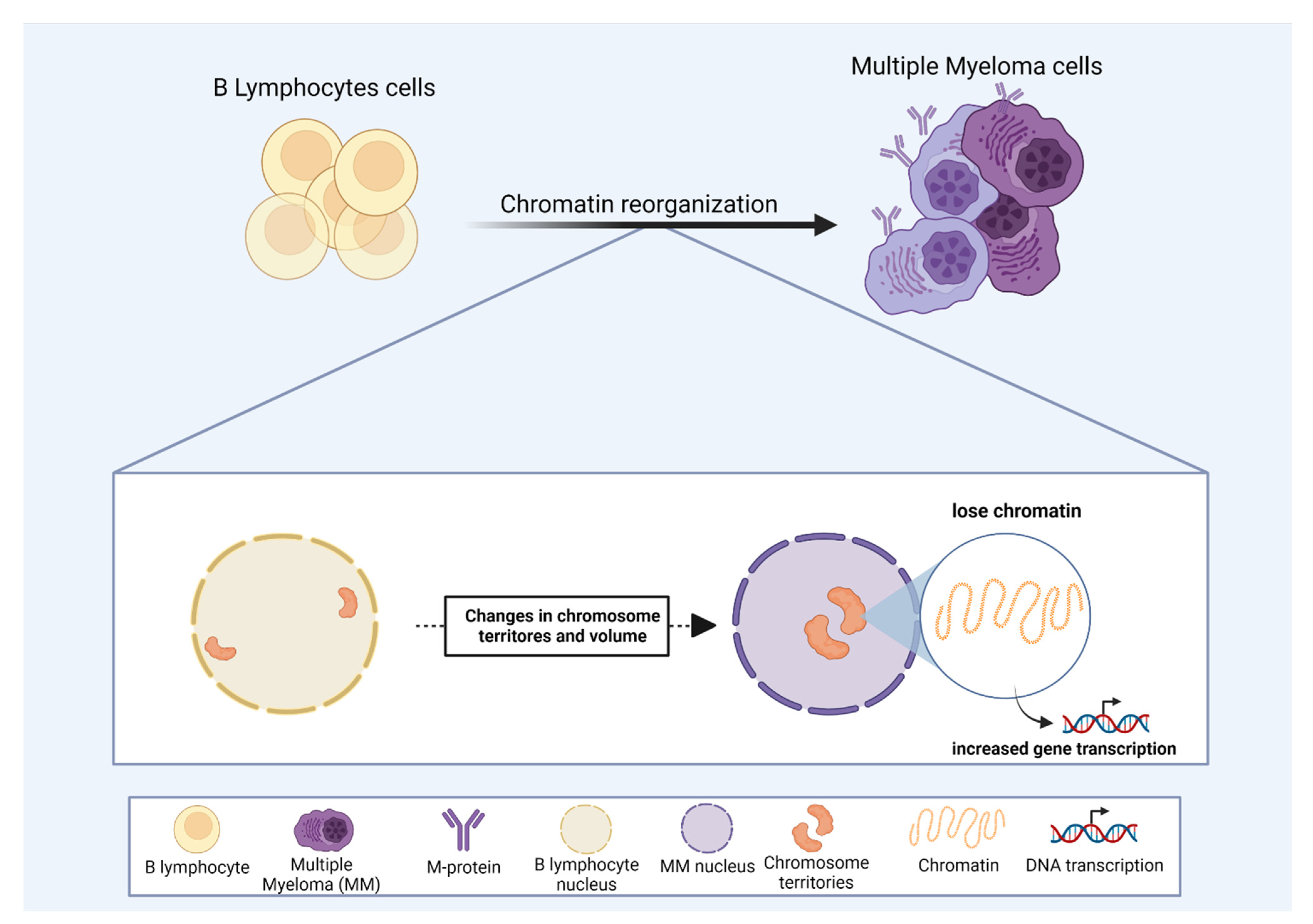
1. Multiple Myeloma
2. Acute Myeloid Leukemia and Secondary Leukemia
3. Radiation Effects on CT in Hematological Malignancies
4. Lymphomas
This entry is adapted from the peer-reviewed paper 10.3390/cells11081368
References
- Fritz, A.J.; Stojkovic, B.; Ding, H.; Xu, J.; Bhattacharya, S.; Gaile, D.; Berezney, R. Wide-scale alterations in interchromosomal organization in breast cancer cells: Defining a network of interacting chromosomes. Hum. Mol. Genet. 2014, 23, 5133–5146.
- Zeitz, M.J.; Marella, N.V.; Malyavantham, K.S.; Goetze, S.; Bode, J.; Raska, I.; Berezney, R. Organization of the amplified type I interferon gene cluster and associated chromosome regions in the interphase nucleus of human osteosarcoma cells. Chromosome Res. 2009, 17, 305–319.
- Marella, N.V.; Bhattacharya, S.; Mukherjee, L.; Xu, J.; Berezney, R. Cell type specific chromosome territory organization in the interphase nucleus of normal and cancer cells. J. Cell. Physiol. 2009, 221, 130–138.
- Parl, F.F.; Dupont, W.D.; Crooke, P.S. Interchromosomal Translocations as a Means to Map Chromosome Territories in Breast Cancer. Cancer Inform. 2019, 18, 117693511984257.
- Feng, L.; Li, H.; Zhang, Y.; Shen, H.; Yu, H.; Zhong, F.; Qin, L.; He, F.; Yang, P.; Tang, Z. Metastasis-related genes in hepatocellular carcinoma cell-lines are clustered on chromosome territories predicted by transcriptome and proteome. Sci. China Chem. 2016, 59, 380–382.
- Schwaederle, M.; Chattopadhyay, R.; Kato, S.; Fanta, P.T.; Banks, K.C.; Choi, I.S.; Piccioni, D.E.; Ikeda, S.; Talasaz, A.; Lanman, R.B.; et al. Genomic Alterations in Circulating Tumor DNA from Diverse Cancer Patients Identified by Next-Generation Sequencing. Cancer Res. 2017, 77, 5419–5427.
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447.
- Bridger, J.M.; Arican-Gotkas, H.D.; Foster, H.A.; Godwin, L.S.; Harvey, A.; Kill, I.R.; Knight, M.; Mehta, I.S.; Ahmed, M.H. The Non-random Repositioning of Whole Chromosomes and Individual Gene Loci in Interphase Nuclei and Its Relevance in Disease, Infection, Aging, and Cancer. Cancer Biol. Nucl. Envel. 2014, 773, 263–279.
- Vargas-Rondón, N.; Villegas, V.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4.
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150.
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418.
- Dimopoulos, M.A.; Terpos, E. Multiple myeloma. Ann. Oncol. 2010, 21, vii143–vii150.
- Palumbo, A.; Anderson, K. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060.
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, Staging, and Management of Multiple Myeloma. Med. Sci. 2021, 9, 3.
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598.
- Abdallah, N.; Rajkumar, S.V.; Greipp, P.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Baughn, L.B.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; et al. Cytogenetic abnormalities in multiple myeloma: Association with disease characteristics and treatment response. Blood Cancer J. 2020, 10, 82.
- Su, C.T.; Chen, L.; Chen, J.; Parkin, B.; Polk, A.; Kandarpa, M.; Cole, C.E.; Campagnaro, E.; Vo, J.; Robinson, D.; et al. Role of Aneuploidy in Transcriptional Regulation and Clinical Prognosis in Relapsed and/or Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 45–46.
- Barwick, B.G.; Neri, P.; Bahlis, N.J.; Nooka, A.K.; Dhodapkar, M.V.; Jaye, D.L.; Hofmeister, C.C.; Kaufman, J.L.; Gupta, V.A.; Auclair, D.; et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat. Commun. 2019, 10, 1911.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348.
- Wang, Y.; Nagarajan, M.; Uhler, C.; Shivashankar, G.V. Orientation and repositioning of chromosomes correlate with cell geometry–dependent gene expression. Mol. Biol. Cell 2017, 28, 1997–2009.
- Schwope, R.; Magris, G.; Miculan, M.; Paparelli, E.; Celii, M.; Tocci, A.; Marroni, F.; Fornasiero, A.; De Paoli, E.; Morgante, M. Open chromatin in grapevine marks candidate CREs and with other chromatin features correlates with gene expression. Plant J. 2021, 107, 1631–1647.
- Jin, Y.; Chen, K.; De Paepe, A.; Hellqvist, E.; Krstic, A.D.; Metang, L.; Gustafsson, C.; Davis, R.E.; Levy, Y.M.; Surapaneni, R.; et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 2018, 131, 2138–2150.
- Ordoñez, R.; Kulis, M.; Russiñol, N.; Chapaprieta, V.; Carrasco-Leon, A.; García-Torre, B.; Charalampopoulou, S.; Clot, G.; Beekman, R.; Meydan, C.; et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020, 30, 1217–1227.
- Sathitruangsak, C.; Righolt, C.H.; Klewes, L.; Tung Chang, D.; Kotb, R.; Mai, S. Distinct and shared three-dimensional chromosome organization patterns in lymphocytes, monoclonal gammopathy of undetermined significance and multiple myeloma. Int. J. Cancer 2017, 140, 400–410.
- Sathitruangsak, C.; Righolt, C.H.; Klewes, L.; Tammur, P.; Ilus, T.; Tamm, A.; Punab, M.; Olujohungbe, A.; Mai, S. Quantitative Superresolution Microscopy Reveals Differences in Nuclear DNA Organization of Multiple Myeloma and Monoclonal Gammopathy of Undetermined Significance. J. Cell. Biochem. 2015, 116, 704–710.
- Broyl, A.; Hose, D.; Lokhorst, H.; de Knegt, Y.; Peeters, J.; Jauch, A.; Bertsch, U.; Buijs, A.; Stevens-Kroef, M.; Beverloo, H.B.; et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 2010, 116, 2543–2553.
- Chen, Y.; Valent, E.T.; Beers, E.H.; Kuiper, R.; Oliva, S.; Haferlach, T.; Chng, W.; Vliet, M.H.; Sonneveld, P.; Larocca, A. Prognostic gene expression analysis in a retrospective, multinational cohort of 155 multiple myeloma patients treated outside clinical trials. Int. J. Lab. Hematol. 2021, 44, 127–134.
- Ezoe, S. Secondary Leukemia Associated with the Anti-Cancer Agent, Etoposide, a Topoisomerase II Inhibitor. Int. J. Environ. Res. Public Health 2012, 9, 2444–2453.
- Reikvam, H.; Hatfield, K.J.; Kittang, A.O.; Hovland, R.; Bruserud, Ø. Acute Myeloid Leukemia with the t(8;21) Translocation: Clinical Consequences and Biological Implications. J. Biomed. Biotechnol. 2011, 2011, 104631.
- Rubtsov, M.A.; Terekhov, S.M.; Razin, S.V.; Iarovaia, O.V. Repositioning of ETO gene in cells treated with VP-16, an inhibitor of DNA-Topoisomerase II. J. Cell. Biochem. 2008, 104, 692–699.
- Rubtsov, M.A.; Glukhov, S.I.; Allinne, J.; Pichugin, A.; Vassetzky, Y.S.; Razin, S.V.; Iarovaia, O.V. Treatment of lymphoid cells with the topoisomerase II poison etoposide leads to an increased juxtaposition of AML1 and ETO genes on the surface of nucleoli. Biopolym. Cell 2011, 27, 398–403.
- Gole, B.; Wiesmüller, L. Leukemogenic rearrangements at the mixed lineage leukemia gene (MLL)—Multiple rather than a single mechanism. Front. Cell Dev. Biol. 2015, 3, 41.
- Super, H.G.; Strissel, P.L.; Sobulo, O.M.; Burian, D.; Reshmi, S.C.; Roe, B.; Zeleznik-Le, N.J.; Diaz, M.O.; Rowley, J.D. Identification of complex genomic breakpoint junctions in the t(9;11) MLL-AF9 fusion gene in acute leukemia. Genes Chromosomes Cancer 1997, 20, 185–195.
- Zhang, Y.; Strissel, P.; Strick, R.; Chen, J.; Nucifora, G.; Le Beau, M.M.; Larson, R.A.; Rowley, J.D. Genomic DNA breakpoints in AML1/RUNX1 and ETO cluster with topoisomerase II DNA cleavage and DNase I hypersensitive sites in t(8;21) leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 3070–3075.
- Glukhov, S.I.; Rubtsov, M.A.; Alexeyevsky, D.A.; Alexeevski, A.V.; Razin, S.V.; Iarovaia, O.V. The Broken MLL Gene Is Frequently Located Outside the Inherent Chromosome Territory in Human Lymphoid Cells Treated with DNA Topoisomerase II Poison Etoposide. PLoS ONE 2013, 8, e75871.
- Huret, J.; Dessen, P.; Bernheim, A. An Atlas on Chromosomes in Hematological Malignancies. Example: 11q23 and MLL partners. Leukemia 2001, 15, 987–989.
- Huret, J.L.; Senon, S.; Bernheim, A.; Dessen, P. An Atlas on genes and chromosomes in oncology and haematology. Cell. Mol. Biol. (Noisy-Le-Grand) 2004, 50, 805–807.
- Gué, M.; Sun, J.-S.; Boudier, T. Simultaneous localization of MLL, AF4 and ENL genes in interphase nuclei by 3D-FISH: MLL translocation revisited. BMC Cancer 2006, 6, 20.
- Soutoglou, E.; Dorn, J.F.; Sengupta, K.; Jasin, M.; Nussenzweig, A.; Ried, T.; Danuser, G.; Misteli, T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007, 9, 675–682.
- Aten, J.A. Dynamics of DNA Double-Strand Breaks Revealed by Clustering of Damaged Chromosome Domains. Science 2004, 303, 92–95.
- Nikiforova, M.N.; Stringer, J.R.; Blough, R.; Medvedovic, M.; Fagin, J.A.; Nikiforov, Y.E. Proximity of Chromosomal Loci That Participate in Radiation-Induced Rearrangements in Human Cells. Science 2000, 290, 138–141.
- Huang, L.; Snyder, A.R.; Morgan, W.F. Radiation-induced genomic instability and its implications for radiation carcinogenesis. Oncogene 2003, 22, 5848–5854.
- Sachs, R.K.; Chen, A.M.; Brenner, D.J. Review: Proximity effects in the production of chromosome aberrations by ionizing radiation. Int. J. Radiat. Biol. 1997, 71, 1–19.
- Lukášová, E.; Kozubek, S.; Kozubek, M.; Kjeronská, J.; Rýznar, L.; Horáková, J.; Krahulcová, E.; Horneck, G. Localisation and distance between ABL and BCR genes in interphase nuclei of bone marrow cells of control donors and patients with chronic myeloid leukaemia. Hum. Genet. 1997, 100, 525–535.
- Kozubek, S.; Lukášová, E.; Rýznar, L.; Kozubek, M.; Lišková, A.; Govorun, R.D.; Krasavin, E.A.; Horneck, G. Distribution of ABL and BCR Genes in Cell Nuclei of Normal and Irradiated Lymphocytes. Blood 1997, 89, 4537–4545.
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234.
- Bártová, E.; Kozubek, S.; Kozubek, M.; Jirsová, P.; Lukášová, E.; Skalníková, M.; Buchníčková, K. The influence of the cell cycle, differentiation and irradiation on the nuclear location of the abl, bcr and c-myc genes in human leukemic cells. Leuk. Res. 2000, 24, 233–241.
- Cafourková, A.; Lukásová, E.; Kozubek, S.; Kozubek, M.; Govorun, R.D.; Koutná, I.; Bártová, E.; Skalníková, M.; Jirsová, P.; Paseková, R.; et al. Exchange aberrations among 11 chromosomes of human lymphocytes induced by γ-rays. Int. J. Radiat. Biol. 2001, 77, 419–429.
- Boei, J.J.W.A.; Fomina, J.; Darroudi, F.; Nagelkerke, N.J.D.; Mullenders, L.H.F. Interphase Chromosome Positioning Affects the Spectrum of Radiation-Induced Chromosomal Aberrations. Radiat. Res. 2006, 166, 319–326.
- Anderson, R.M.; Stevens, D.L.; Goodhead, D.T. M-FISH analysis shows that complex chromosome aberrations induced by -particle tracks are cumulative products of localized rearrangements. Proc. Natl. Acad. Sci. USA 2002, 99, 12167–12172.
- Anderson, R.M.; Papworth, D.G.; Stevens, D.L.; Sumption, N.D.; Goodhead, D.T. Increased complexity of radiation-induced chromosome aberrations consistent with a mechanism of sequential formation. Cytogenet. Genome Res. 2006, 112, 35–44.
- Balajee, A.S.; Sanders, J.T.; Golloshi, R.; Shuryak, I.; McCord, R.P.; Dainiak, N. Investigation of Spatial Organization of Chromosome Territories in Chromosome Exchange Aberrations after Ionizing Radiation Exposure. Health Phys. 2018, 115, 77–89.
- Küppers, R.; Rajewsky, K. The origin of Hodgkin and Reed/Sternberg cells in Hodgkin’s disease. Annu. Rev. Immunol. 1998, 16, 471–493.
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390.
- Guffei, A.; Sarkar, R.; Klewes, L.; Righolt, C.; Knecht, H.; Mai, S. Dynamic chromosomal rearrangements in Hodgkin’s lymphoma are due to ongoing three-dimensional nuclear remodeling and breakage-bridge-fusion cycles. Haematologica 2010, 95, 2038–2046.
- Knecht, H.; Sawan, B.; Lichtensztejn, D.; Lemieux, B.; Wellinger, R.J.; Mai, S. The 3D nuclear organization of telomeres marks the transition from Hodgkin to Reed–Sternberg cells. Leukemia 2009, 23, 565–573.
- Righolt, C.H.; Knecht, H.; Mai, S. DNA Superresolution Structure of Reed-Sternberg Cells Differs Between Long-Lasting Remission Versus Relapsing Hodgkin’s Lymphoma Patients. J. Cell. Biochem. 2016, 117, 1633–1637.
- Roix, J.J.; McQueen, P.G.; Munson, P.J.; Parada, L.A.; Misteli, T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 2003, 34, 287–291.
- Germini, D.; Tsfasman, T.; Klibi, M.; El-Amine, R.; Pichugin, A.; Iarovaia, O.V.; Bilhou-Nabera, C.; Subra, F.; Bou Saada, Y.; Sukhanova, A.; et al. HIV Tat induces a prolonged MYC relocalization next to IGH in circulating B-cells. Leukemia 2017, 31, 2515–2522.
- Hapgood, G.; Savage, K.J. The biology and management of systemic anaplastic large cell lymphoma. Blood 2015, 126, 17–25.
- Mathas, S.; Kreher, S.; Meaburn, K.J.; Johrens, K.; Lamprecht, B.; Assaf, C.; Sterry, W.; Kadin, M.E.; Daibata, M.; Joos, S.; et al. Gene deregulation and spatial genome reorganization near breakpoints prior to formation of translocations in anaplastic large cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 5831–5836.
- Klein, I.A.; Resch, W.; Jankovic, M.; Oliveira, T.; Yamane, A.; Nakahashi, H.; Di Virgilio, M.; Bothmer, A.; Nussenzweig, A.; Robbiani, D.F.; et al. Translocation-Capture Sequencing Reveals the Extent and Nature of Chromosomal Rearrangements in B Lymphocytes. Cell 2011, 147, 95–106.
- Steininger, A.; Ebert, G.; Becker, B.V.; Assaf, C.; Möbs, M.; Schmidt, C.A.; Grabarczyk, P.; Jensen, L.R.; Przybylski, G.K.; Port, M.; et al. Genome-Wide Analysis of Interchromosomal Interaction Probabilities Reveals Chained Translocations and Overrepresentation of Translocation Breakpoints in Genes in a Cutaneous T-Cell Lymphoma Cell Line. Front. Oncol. 2018, 8, 183.
- Branco, M.R.; Pombo, A. Intermingling of Chromosome Territories in Interphase Suggests Role in Translocations and Transcription-Dependent Associations. PLoS Biol. 2006, 4, e138.
- Tavares-Cadete, F.; Norouzi, D.; Dekker, B.; Liu, Y.; Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nat. Struct. Mol. Biol. 2020, 27, 1105–1114.