CCAAT/enhancer-binding protein δ (C/EBPδ) is a transcription factor involved in growth arrest and differentiation, which has consequently been suggested to harbor tumor suppressive activities. However, C/EBPδ over-expression also correlates with poor prognosis in glioblastoma and promotes genomic instability in cervical cancer, hinting at an oncogenic role of C/EBPδ in these contexts. This entry outlines the role of C/EBPδ in pancreatic ductal adenocarcinoma. In cell lines of this cancer, C/EBPδ exerts a tumor suppressive role by attenuating clonogenicity, proliferation and tumor sphere formation.
- CCAAT/Enhancer-Binding Protein Delta
- Pancreatic Ductal Adenocarcinoma
- Tumor Suppressor
1. Introduction
1.1. Pancreatic ductal adenocarcinoma
Pancreatic cancer is a devastating disease with a survival outcome that is the worst of all human cancers[1]. The 5-year survival rate upon diagnosis is a little over 9% and overall mortality reaches 99%[2][3]. Due to the late onset of symptoms, only 15–20% of patients present with resectable disease, whereas the remaining patients present with metastatic or locally advanced disease, which cannot be resected. The median survival of the selected group of resectable patients, however, increases only to around 23 months whereas 5-year survival rates remain below 20%[4][5][6]. Eventually, the majority of these patients with a resectable primary tumor will succumb due to metastatic disease as well[7]. Thus, there is a clear clinical need to better understand the processes that drive pancreatic cancer and guide the development of novel avenues for rational treatment of this disease.
Only a limited number of tumor suppressor genes have been formally established in pancreatic ductal adenocarcinoma. Mutations in genes such as TP53, SMAD4, PTEN, and CDKN2A are present in over 70% of pancreatic ductal adenocarcinomas, and mutations in these tumor suppressors are well known to drive tumor progression. As opposed to their clear biological relevance, mutations in tumor suppressor genes typically are of limited therapeutic value[12][13]. To improve patient treatment, the identification of tumor suppressor genes that could serve as therapeutic targets is therefore eagerly awaited.
1.1. Roles of C/EBPδ in tumorigenesis
CCAAT/enhancer-binding protein δ (C/EBPδ) is a member of the C/EBP superfamily of transcription factors, which consists of six unique members (α, β, γ, δ, ε and ζ)[14]. Soon after its discovery, C/EBPδ was implied to act as a tumor suppressor by inducing growth arrest and differentiation in breast cancer[15]. Indeed, C/EBPδ expression promotes CDC27 expression, leading to increased degradation of the cell cycle proteins cyclin D1, cyclin B1, Plk-1, and Skp2[16]. Furthermore, expression of C/EBPδ is associated with downregulation of c-Myc and cyclin E, and upregulation of the cyclin-dependent kinase inhibitor p27 in the leukemia cell lines K562 and KCL22, leading to growth arrest and differentiation[17]. In A431 cervical cancer cells, C/EBPδ expression leads to the induction of apoptosis via the transcriptional regulation of the pro-apoptotic genes PPARG2 and GADD153[18]. Moreover, C/EBPδ is involved in the regulation of pro-apoptotic gene expression and growth arrest during mammary gland involution[19][20]. In line with these data, C/EBPδ indeed acts as a tumor suppressor in breast cancer[21][22][23], ovarian serous carcinoma[24], cervical carcinoma[25], leukemia[26] and hepatocellular carcinoma[27][28].
In contrast to the presumed tumor suppressor role of C/EBPδ, several studies suggest that C/EBPδ may actually drive tumor progression in certain cancers. Indeed, C/EBPδ over-expression correlates with poor prognosis in glioblastoma[29]; it is required for efficient metastatic growth of mammary tumors[30], and drives proliferation and invasiveness of urothelial carcinoma cells, thereby driving metastatic disease leading to a reduced disease-specific survival[31]. Finally, C/EBPδ promotes tumorigenesis in the cervix by inducing aneuploidy and centromere abnormalities[32].
2. C/EBPδ in Pancreatic Ductal Adenocarcinoma
2.1. C/EBPδ is lost in PDAC and correlates with N-status and survival
Data mining of publicly available microarray datasets and immunohistochemical analysis of in-house tissue microarrays showed that CEBPD gene and protein expression are significantly decreased in pancreatic ductal adenocarcinomas versus healthy pancreatic tissue. Hereby, healthy ductal cells express high nuclear levels of C/EBPδ which is at odds with the general notion that C/EBPδ expression is typically low under normal conditions [47]. More importantly, C/EBPδ levels are dramatically reduced in ductal adenocarcinoma cells as compared to normal ductal cells. Next to that, C/EBPδ is negatively correlated to N-status and patient survival in this patient cohort (Figure 1). As C/EBPδ induces growth arrest[16][17], it is tempting to speculate that the loss of C/EBPδ conversely facilitates tumorigenesis. Such a role of C/EBPδ would be in line with previous studies showing that C/EBPδ is a tumor suppressor in leukemia, breast cancer, hepatocellular carcinoma and cervical cancer [21][22][23][24][25][26][27][28].
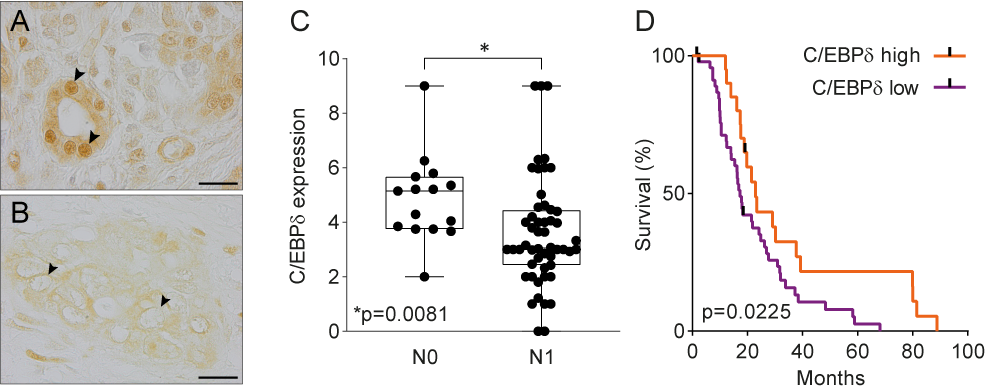
Figure 1. (A) Normal pancreas duct cells express high levels of C/EBPδ protein. Scale bar is 20 µm. (B) C/EBPδ is lost in PDAC tissue samples. (C) C/EBPδ is significantly lower expressed in samples of patients with lymph node involvement than in patients without. (D) High C/EBPδ-expression correlates with improved survival in PDAC patients.
2.2. C/EBPδ reduces PDAC cell tumorigenicity in vitro
C/EBPδ expression is low in MIA PaCa-2 and PANC-1, two commonly used pancreatic ductal adenocarcinoma cell lines. This is in line with observations in pancreatic cancer patients and with a potential role of C/EBPδ as a tumor suppressor in pancreatic cancer. More importantly, their low C/EBPδ expression makes these cells suitable model systems for C/EBPδ re-expression studies with the assumption that rescuing C/EBPδ expression would reduce their tumorigenic capacity in case C/EBPδ acts as a genuine tumor suppressor. PANC-1 and MIA PaCa-2 cells indeed showed decreased clonogenicity and proliferation upon C/EBPδ re-expression (Figure 2 A-B). Additionally, these effects were dose dependent, implying a relation between tumorigenicity and C/EBPδ expression levels. Conversely, shRNA-mediated silencing of C/EBPδ enhances proliferation in a dose-dependent manner strongly suggesting that C/EBPδ expression levels negatively correlate with the proliferative capacity of pancreatic ductal adenocarcinoma cells.
Next to driving proliferation and clonogenicity of pancreatic ductal adenocarcinoma cells, loss of C/EBPδ also seems to promote anchorage-independent growth. This clonogenic capacity in a three-dimensional, anchorage-free environment is considered a key hallmark of oncogenic transformation and is considered the most accurate and stringent in vitro assay for detecting malignant transformation of cells. We found a marked reduction in the number of spheres formed by MIA PaCa-2 and PANC-1 cells upon re-expression of C/EBPδ (Figure 2C). Interestingly, although C/EBPδ re-expression limits sphere formation in these cell lines, PANC-1 cells appear to be more susceptible to reversing oncogenic properties upon C/EBPδ induction. Although the precise mechanism underlying the reduction in anchorage-independent growth of pancreatic adenocarcinoma cancer cell remains to be established, it is tempting to speculate that these data explain the correlation found between C/EBPδ expression and lymph node invasion observed in our patient cohort. Indeed, anchorage-independent growth is strongly associated with the metastatic potential of cancer cells[44]. Irrespective of the actual mechanism, the reduced overall survival in pancreatic ductal adenocarcinoma patients with low C/EBPδ levels might be directly linked to the increased lymph node metastases in these patients.
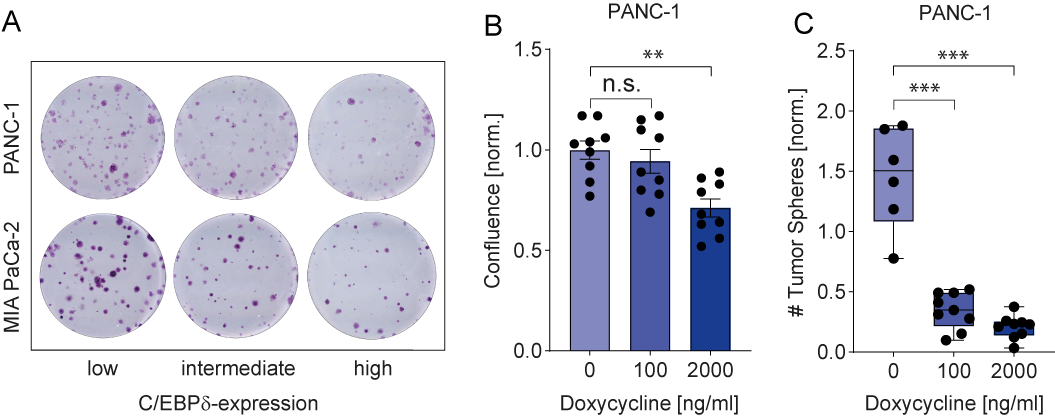
Figure 2. (A) Crystal violet stainings show that re-expression of C/EBPδ reduces the clonogenicity of two PDAC cell lines in a dose-dependent manner. (B) A doxycycline-inducible system for C/EBPδ re-expression shows hampered proliferation and (C) decreased tumor sphere formation upon induction in a dose-dependent manner.
Taken together, these results point towards a clear direction where C/EBPδ regulates the proliferation and clonogenic capacities of PDAC cells. However, as two-dimensional monoculture experiments in vitro cannot account for important factors, such as stromal and immune infiltration, in vivo validation of these findings is urgently needed to manifest the notion that C/EBPδ might act as a tumor suppressor in PDAC.
2.3. C/EBPδ - a promising target in PDAC treatment?
C/EBPδ is obviously not the first tumor suppressor identified in pancreatic adenocarcinoma. Indeed, genes such as TP53, SMAD4, PTEN, and CDKN2A are well-known tumor suppressors and mutations in these genes, which are present in over 70% of pancreatic ductal adenocarcinomas, drive tumor progression. As opposed to the other classical tumor suppressors, C/EBPδ seems, however, neither hypermethylated, nor mutated, lost or deleted in pancreatic adenocarcinoma (re-analysis of previously published data[48][49][50]) suggesting re-activation of C/EBPδ may hold therapeutic promise in the setting of pancreatic adenocarcinoma.
The mechanism via which C/EBPδ exerts its tumor suppressive and anti-metastatic effects remains elusive. In an attempt to uncover the underlying mechanism, the expression of different putative targets of C/EBPδ involved in cell cycle progression, apoptosis, stemness and the leading-edge genes of the above described gene set enrichment analyses (GSEAs) have been investigated. Enhanced expression of CDKN1A (p21) along with suppressed cyclin-dependent kinases CDK1, CDK2 and CDK6 point towards cell cycle arrest as a main mechanism of the observed C/EBPδ-induced effects in PDAC cells. Interestingly, however, C/EBPδ also appears to affect almost all of the other investigated pathways to some degree. Hence, the data do not yet allow firm conclusions on the downstream pathways affected by C/EBPδ.
To date, several agents have been described to effectively induce C/EBPδ expression. Among these activators are interleukin-6, which elicited growth-inhibiting effects on LNCaP prostate cancer cells via C/EBPδ activation[51], 1-(2-hydroxy-5-methylphenyl)-3-phenyl-1, 3-propanedione (HMDB), which attenuated the growth of A431 epidermoid carcinoma xenografts in severe combined immunodeficient mice[18][27], and metformin, which induced autophagy of Huh7 liver cancer cells via C/EBPδ activation[52]. Next to that, C/EBPδ has been induced by various external stimuli in the inflammatory context. In the context of PDAC, further studies are needed to find potent upstream regulators of C/EBPδ.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12092546
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Stark, A.P.; Sacks, G.D.; Rochefort, M.M.; Donahue, T.R.; Reber, H.A.; Tomslinson, J.S.; Dawson, D.W.; Eibl, G.; Hines, O.J. Long-term Survival in Patients with Pancreatic Ductal Adenocarcinoma. Surgery 2016, 159, 1520–1527. [Google Scholar] [CrossRef]
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, e102655. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Christenson, E.S.; Jaffee, E.; Azad, N.S. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: A bright future. Lancet Oncol. 2020, 21, e135–e145. [Google Scholar] [CrossRef]
- Cardenes, H.R.; Chiorean, E.G.; Dewitt, J.; Schmidt, M.; Loehrer, P. Locally advanced pancreatic cancer: Current therapeutic approach. Oncologist 2006, 11, 612–623. [Google Scholar] [CrossRef]
- Gharibi, A.; Adamian, Y.; Kelber, J.A. Cellular and molecular aspects of pancreatic cancer. Acta. Histochem. 2016, 118, 305–316. [Google Scholar] [CrossRef]
- Ahn, D.H.; Bekaii-Saab, T. Ampullary cancer: An overview. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, 112–115. [Google Scholar] [CrossRef]
- Freeny, P.C. Computed tomography in the diagnosis and staging of cholangiocarcinoma and pancreatic carcinoma. Ann. Oncol. 1999, 10, 12–17. [Google Scholar] [CrossRef]
- Hrad, V.; Abebe, Y.; Ali, S.H.; Velgersdyk, J.; Al Hallak, M.; Imam, M. Risk and Surveillance of Cancers in Primary Biliary Tract Disease. Gastroenterol. Res. Pract. 2016, 2016, e3432640. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, e42. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Elpek, K.G.; Vinjamoori, A.; Zimmerman, S.M.; Chu, G.C.; Yan, H.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kB-cytokine network. Cancer Discov. 2011, 1, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef]
- Sivko, G.S.; DeWille, J.W. CCAAT/enhancer binding protein delta (C/EBP delta) regulation and expression in human mammary epithelial cells: I. “Loss of function” alterations in the C/EBP delta growth inhibitory pathway in breast cancer cell lines. J. Cell. Biochem. 2004, 93, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Pawar, S.A.; Sarkar, T.R.; Balamurugan, K.; Sharan, S.; Wang, J.; Zhang, Y.; Dowdy, S.F.; Huang, A.M.; Sterneck, E. C/EBPdelta targets cyclin D1 for proteasome-mediated degradation via induction of CDC27/APC3 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 9210–9215. [Google Scholar] [CrossRef]
- Gery, S.; Tanosaki, S.; Hofmann, W.K.; Koppel, A.; Koeffler, H.P. C/EBPdelta expression in a BCR-ABL-positive cell line induces growth arrest and myeloid differentiation. Oncogene 2005, 24, 1589–1597. [Google Scholar] [CrossRef]
- Pan, Y.C.; Li, C.F.; Ko, C.Y.; Pan, M.H.; Chen, P.J.; Tseng, J.T.; Wu, W.C.; Chang, W.C.; Huang, A.M.; Sterneck, E.; et al. CEBPD reverses RB/E2F1-mediated gene repression and participates in HMDB-induced apoptosis of cancer cells. Clin. Cancer Res. 2010, 16, 5770–5780. [Google Scholar] [CrossRef]
- O’Rourke, J.; Yuan, R.; DeWille, J. CCAAT/enhancer-binding protein-delta (C/EBP-delta) is induced in growth-arrested mouse mammary epithelial cells. J. Biol. Chem. 1997, 272, 6291–6296. [Google Scholar] [CrossRef]
- Thangaraju, M.; Rudelius, M.; Bierie, B.; Raffeld, M.; Sharan, S.; Hennighausen, L.; Huang, A.M.; Sterneck, E. C/EBPdelta is a crucial regulator of pro-apoptotic gene expression during mammary gland involution. Development 2005, 132, 4675–4685. [Google Scholar] [CrossRef]
- Mendoza-Villanueva, D.; Balamurugan, K.; Ali, H.R.; Kim, S.R.; Sharan, S.; Johnson, R.C.; Merchant, A.S.; Caldas, C.; Landberg, G.; Sterneck, E. The C/EBPδ protein is stabilized by estrogen receptor α activity, inhibits SNAI2 expression and associates with good prognosis in breast cancer. Oncogene 2016, 35, 6166–6176. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, C.; Monteverde, M.; Lattanzio, L.; Gojis, O.; Rudraraju, B.; Fortunato, M.; Syed, N.; Thompson, A.; Garrone, O.; Merlano, M.; et al. Site-specific CpG methylation in the CCAAT/enhancer binding protein delta (CEBPδ) CpG island in breast cancer is associated with metastatic relapse. Br. J. Cancer 2012, 107, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Sivko, G.S.; DeWille, J.W. Promoter methylation reduces C/EBPdelta (CEBPD) gene expression in the SUM-52PE human breast cancer cell line and in primary breast tumors. Breast Cancer Res. Treat. 2006, 95, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Sowamber, R.; Chehade, R.; Bitar, M.; Dodds, L.V.; Milea, A.; Slomovitz, B.; Shaw, P.A.; George, S.H.L. CCAAT/enhancer binding protein delta (C/EBPδ) demonstrates a dichotomous role in tumor initiation and promotion of epithelial carcinoma. EBioMedicine 2019, 44, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.Y.; Hsu, H.C.; Shen, M.R.; Chang, W.C.; Wang, J.M. Epigenetic silencing of CCAAT/enhancer-binding protein delta activity by YY1/polycomb group/DNA methyltransferase complex. J. Biol. Chem. 2008, 283, 30919–33032. [Google Scholar] [CrossRef]
- Agrawal, S.; Hofmann, W.K.; Tidow, N.; Ehrich, M.; Van den Boom, D.; Koschmieder, S.; Berdel, W.E.; Serve, H.; Müller-Tidow, C. The C/EBPdelta tumor suppressor is silenced by hypermethylation in acute myeloid leukemia. Blood 2007, 109, 3895–3905. [Google Scholar] [CrossRef]
- Li, C.F.; Tsai, H.H.; Ko, C.Y.; Pan, Y.C.; Yen, C.J.; Lai, H.Y.; Yuh, C.H.; Wu, W.C.; Wang, J.M. HMDB and 5-AzadC Combination Reverses Tumor Suppressor CCAAT/Enhancer-Binding Protein Delta to Strengthen the Death of Liver Cancer Cells. Mol. Cancer Ther. 2015, 14, 2623–2633. [Google Scholar] [CrossRef]
- Liu, P.; Cao, W.; Ma, B.; Li, M.; Chen, K.; Sideras, K.; Duitman, J.W.; Sprengers, D.; Khe Tran, T.C.; Ijzermans, J.N.M.; et al. Action and clinical significance of CCAAT/enhancer-binding protein delta in hepatocellular carcinoma. Carcinogenesis 2019, 40, 155–163. [Google Scholar] [CrossRef]
- Cooper, L.A.; Gutman, D.A.; Chisolm, C.; Appin, C.; Kong, J.; Rong, Y.; Kurc, T.; Van Meir, E.G.; Saltz, J.H.; Moreno, C.S.; et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am. J. Pathol. 2012, 180, 2108–2119. [Google Scholar] [CrossRef]
- Balamurugan, K.; Wang, J.; Tsai, H.; Sharan, S.; Anver, A.; Leighty, R.; Sterneck, E. The tumour suppressor C/EBPδ inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J. 2010, 29, 4106–4117. [Google Scholar] [CrossRef]
- Wang, Y.H.; Wu, W.J.; Wang, W.J.; Huang, H.Y.; Li, W.M.; Yeh, B.W.; Wu, T.F.; Shiue, Y.L.; Sheu, J.J.; Wang, J.M.; et al. CEBPD amplification and overexpression in urothelial carcinoma: A driver of tumor metastasis indicating adverse prognosis. Oncotarget 2015, 6, 31069–31084. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.R.; Li, C.F.; Hung, L.Y.; Huang, A.M.; Tseng, J.T.; Tsou, J.H.; Wang, J.M. CCAAT/enhancer-binding protein delta mediates tumor necrosis factor alpha-induced Aurora kinase C transcription and promotes genomic instability. J. Biol. Chem. 2011, 286, 28662–28670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Schetter, A.; He, P.; Funamizu, N.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Hassan, R.; Yfantis, H.G.; Lee, D.H.; et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e31507. [Google Scholar] [CrossRef]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.R.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009, 16, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Villanueva, A.; Hoshida, Y.; Peix, J.; Nevell, P.; Minguez, B.; LeBlanc, A.C.; Donovan, D.J.; Thung, S.N.; Sole, M.; et al. Focal Gains of Vascular Endothelial Growth Factor A and Molecular Classification of Hepatocellular Carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, F.; Chang, T.; Wang, T.; Hsu, T.; Chi, J.; Hsiao, Y.; Li, C.; Wang, J. Hepatoma-derived growth factor supports the antiapoptosis and profibrosis of pancreatic stellate cells. Cancer Lett. 2019, 457, 180–190. [Google Scholar] [CrossRef]
- Badea, L.; Herlea, V.; Dima, S.O.; Dumitrascu, T.; Popescu, I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepato Gastroenterol. 2008, 55, 2016–2027. [Google Scholar]
- Grimont, A.; Pinho, A.V.; Cowley, M.J.; Augereau, C.; Mawson, A.; Giry-Laterriere, M.; Van den Steen, G.; Waddell, N.; Pajic, M.; Sempoux, C.; et al. SOX9 regulates ERBB signalling in pancreatic cancer development. Gut 2015, 64, 1790–1799. [Google Scholar] [CrossRef]
- Raphael, B.J.; Aguirre, A.J. Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Yang, S.; He, P.; Wang, J.; Schetter, A.; Tang, W.; Funamizu, N.; Yanaga, K.; Uwagawa, T.; Satoskar, A.R.; Gaedcke, J.; et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res. 2016, 76, 3838–3850. [Google Scholar] [CrossRef] [PubMed]
- Dijk, F.; Veenstra, V.L.; Soer, E.C.; Dings, M.P.G.; Zhao, L.; Halfwerk, J.B.; Hooijer, G.K.; Damhofer, H.; Marzano, M.; Steins, A.; et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020, 10, e337. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmordgoli, M.; Martinez, E.; Vegesna, R.; Hoon, K.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.w.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, e2612. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2790–2805. [Google Scholar] [CrossRef] [PubMed]
- Maupin, K.A.; Sinha, A.; Eugster, E.; Miller, J.; Ross, J.; Paulino, V.; Keshamouni, V.G.; Tran, N.; Berens, M.; Webb, C.; et al. Glycogene expression alterations associated with pancreatic cancer epithelial-mesenchymal transition in complementary model systems. PLoS ONE 2010, 5, e13002. [Google Scholar] [CrossRef] [PubMed]
- R2: Genomics Analysis and Visualization Platform. Available online: http://r2.amc.nl (accessed on 5 August 2020).
- Balamurugan, K.; Sterneck, E. The many faces of C/EBPδ and their relevance for inflammation and cancer. Int. J. Biol. Sci. 2013, 9, 917–933. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Dis. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 269. [Google Scholar] [CrossRef]
- Broad Institute; Firehose Broad GDAC; PAAD. Available online: https://gdac.broadinstitute.org/ (accessed on 3 December 2019).
- DeWille, J.W.; Sanford, D.C. C/EBPdelta is a downstream mediator of IL-6 induced growth inhibition of prostate cancer cells. Prostate 2005, 63, 143–154. [Google Scholar] [CrossRef]
- Tsai, H.H.; Lai, H.Y.; Chen, Y.C.; Li, C.F.; Huang, H.S.; Liu, H.S.; Tsai, Y.S.; Wang, J.M. Metformin promotes apoptosis in hepatocellular carcinoma through the CEBPD-induced autopathy pathway. Oncotarget 2017, 8, 13832–13845.