Interferon regulatory factors (IRFs), a family of transcription factors that regulate Interferon (IFN) expression, play important roles in both innate and adaptive immune responses. IRF-involved signaling pathways contribute to hepatic inflammation, insulin resistance, and immune cell activation, such as macrophage polarization, playing critical roles in non-alcoholic fatty liver disease (NAFLD) or non-alcoholic steatohepatitis (NASH) pathogenesis. Treatments such as microRNAs, PPAR modulators, anti-inflammatory agents, and TLR agonists or antagonists that modulate IRF-mediated signaling pathways can ameliorate the progression of NAFLD to NASH.
- non-alcoholic fatty liver disease
- non-alcoholic steatohepatitis
- interferon
- interferon regulatory factor
1. Introduction
2. Interferons in Non-Alcoholic Fatty Liver Disease (NAFLD) and Non-Alcoholic Steatohepatitis (NASH)
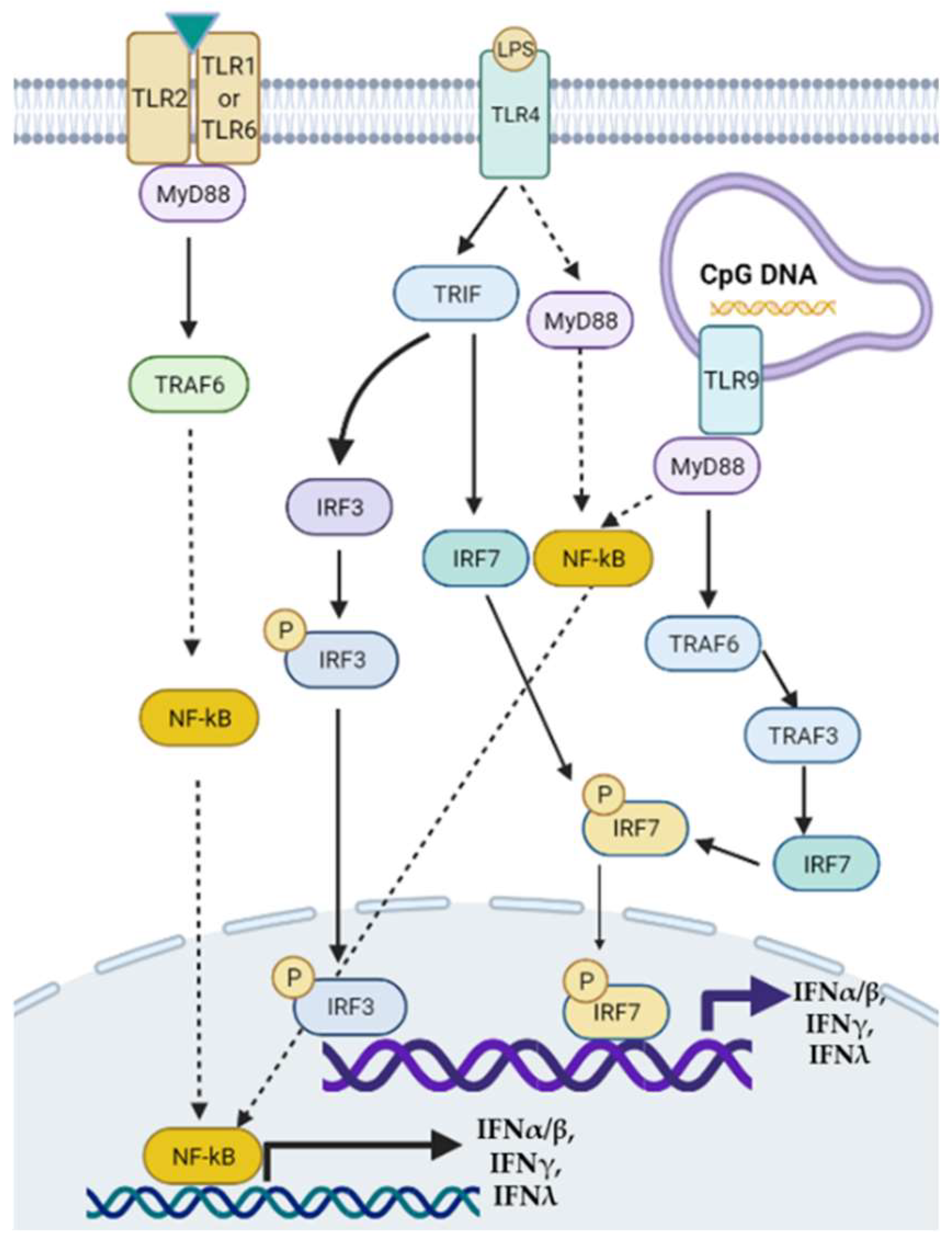 Figure 1. Toll-like receptor-mediated signaling pathways of interferon (IFN) transcription. Modulation of TLRs such as TLR2, TLR4, and TLR9 can activate downstream MyD88/NF-κB, MyD88/TRAF6/3/IRF, or TRIF/IRF3/7 signaling pathways to regulate the transcription of type I, II, and III IFNs. TRAF: tumor necrosis factor receptor-associated factor.
Figure 1. Toll-like receptor-mediated signaling pathways of interferon (IFN) transcription. Modulation of TLRs such as TLR2, TLR4, and TLR9 can activate downstream MyD88/NF-κB, MyD88/TRAF6/3/IRF, or TRIF/IRF3/7 signaling pathways to regulate the transcription of type I, II, and III IFNs. TRAF: tumor necrosis factor receptor-associated factor.3. Roles of IRFs in NAFLD and NASH
3.1. IRF1
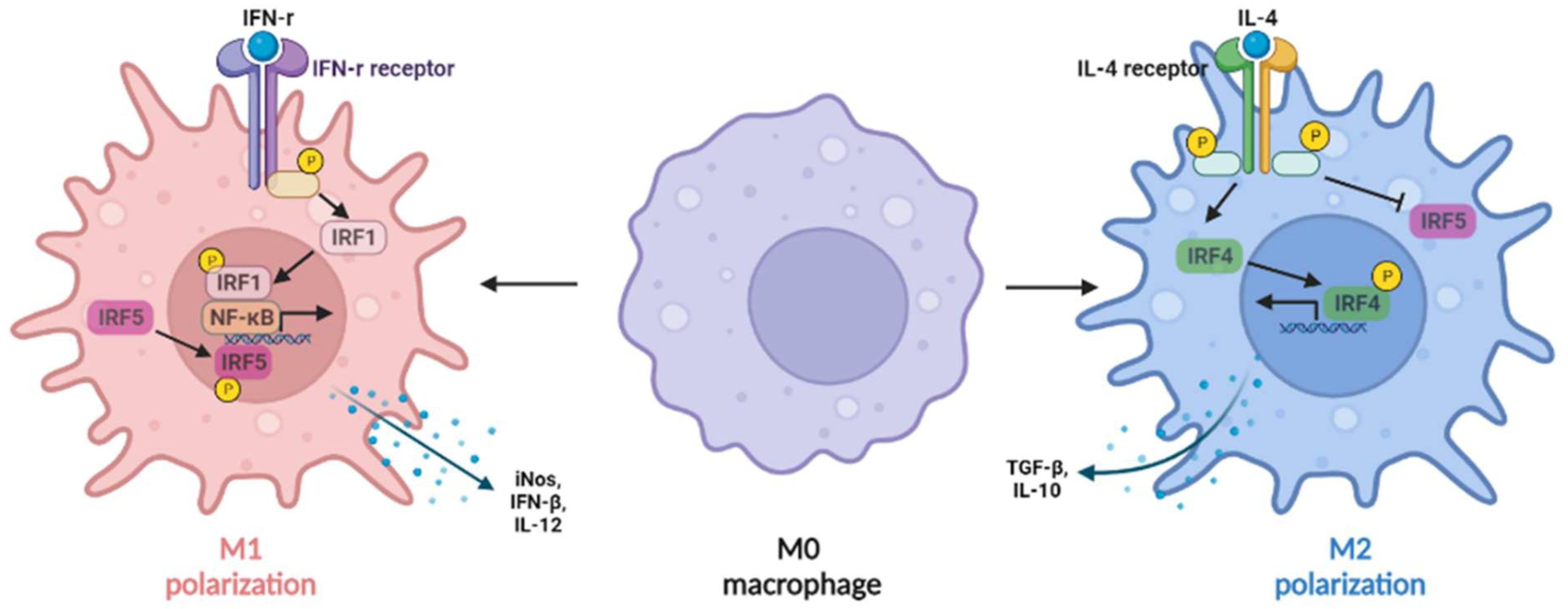 Figure 2. IRFs regulate macrophage polarization. The expression of IRF1 in macrophages can be induced by IFN-γ, synergizing with nuclear factor kappa B (NF-κB) to induce IL-12, inducible nitric oxide synthase (iNOS), and IFN-β production (M1-like macrophages), whereas IL-4 can induce M2-like macrophage polarization by activating IRF4 signaling to prevent IRF5-mediated M1 polarization.
Figure 2. IRFs regulate macrophage polarization. The expression of IRF1 in macrophages can be induced by IFN-γ, synergizing with nuclear factor kappa B (NF-κB) to induce IL-12, inducible nitric oxide synthase (iNOS), and IFN-β production (M1-like macrophages), whereas IL-4 can induce M2-like macrophage polarization by activating IRF4 signaling to prevent IRF5-mediated M1 polarization.3.2. IRF2
3.3. IRF3
3.4. IRF4
3.5. IRF5
3.6. IRF6
3.7. IRF7
3.8. IRF8
3.9. IRF9
| IRFs | Model | Expression * | Function | References |
|---|---|---|---|---|
| IRF1 | NASH rat | Increased | IFN-γ in rat NASH liver upregulated IRF1 expression, resulting in liver inflammation progression. | [11] |
| IRF2 | M1-like macrophages | Decreased | Knockdown of IRF2 accelerated lipopolysaccharide (LPS)-induced activation of macrophages by regulating hypoxia-inducible factor 1-alpha (HIF-1α)-dependent glycolysis. | [44] |
| IRF3 | NAFLD mice | Decreased | IRF3 deficiency dramatically promoted diet-induced hepatic steatosis, and insulin resistance, whereas overexpression of IRF3 induced hemostasis of glucose and lipid balance metabolism, via regulating nuclear factor-kappa B kinase subunit beta (IKKβ)/nuclear factor kappa B (NF-κB) signaling pathway. | [46] |
| IRF4 | M2-like macrophages | Increased | IRF4 is involved in M2-like macrophage polarization induced by IL-4 or mediated by the mTORC2 signaling pathway. | [48][49] |
| IRF5 | Mice with liver fibrosis | Increased | Mice with IRF5 knockdown in myeloid cells were protected from metabolic stress or toxin-induced liver fibrosis, compared with wild-type controls. | [53] |
| IRF6 | NAFLD mice | Decreased | Cellular mechanism study showed that knockout IRF6 specifically in hepatocytes accelerated liver steatosis, while overexpression of IRF6 in hepatocytes ameliorated liver steatosis. | [54] |
| IRF7 | Obese mice | Increased | IRF7 deficiency reduced body weight, insulin resistance, hepatic macrophage infiltration, inflammation, and steatosis in mice on a high-fat diet (HFD). | [55] |
| IRF8 | Zebrafish with liver fibrosis | Increased | Knocking down IRF8 in zebrafish caused a reduction in macrophage numbers and the number and activation of hepatic stellate cells. | [60][61] |
| IRF9 | Obese mice | Decreased | IRF9 knockout increased insulin resistance, hepatic steatosis, inflammation in mice on HFD. | [63] |
This entry is adapted from the peer-reviewed paper 10.3390/gastroent13020016
References
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133.
- Zheng, Q.; Martin, R.C.; Shi, X.; Pandit, H.; Yu, Y.; Liu, X.; Guo, W.; Tan, M.; Bai, O.; Meng, X.; et al. Lack of FGF21 promotes NASH-HCC transition via hepatocyte-TLR4-IL-17A signaling. Theranostics 2020, 10, 9923–9936.
- Hindson, J. Molecular landscape of NASH-HCC. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 456.
- Zhang, C.; Yang, M. The Emerging Factors and Treatment Options for NAFLD-Related Hepatocellular Carcinoma. Cancers 2021, 13, 3740.
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533–543.
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075.
- Alwahsh, S.M.; Gebhardt, R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch. Toxicol. 2017, 91, 1545–1563.
- Alwahsh, S.M.; Xu, M.; Seyhan, H.A.; Ahmad, S.; Mihm, S.; Ramadori, G.; Schultze, F.C. Diet high in fructose leads to an overexpression of lipocalin-2 in rat fatty liver. World J. Gastroenterol. 2014, 20, 1807–1821.
- Luo, X.Y.; Takahara, T.; Kawai, K.; Fujino, M.; Sugiyama, T.; Tsuneyama, K.; Tsukada, K.; Nakae, S.; Zhong, L.; Li, X.K. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G891–G899.
- Choi, W.M.; Ryu, T.; Lee, J.H.; Shim, Y.R.; Kim, M.H.; Kim, H.H.; Kim, Y.E.; Yang, K.; Kim, K.; Choi, S.E.; et al. Metabotropic Glutamate Receptor 5 in Natural Killer Cells Attenuates Liver Fibrosis by Exerting Cytotoxicity to Activated Stellate Cells. Hepatology 2021, 74, 2170–2185.
- Li, J.; Chen, Q.; Yi, J.; Lan, X.; Lu, K.; Du, X.; Guo, Z.; Guo, Y.; Geng, M.; Li, D.; et al. IFN-γ contributes to the hepatic inflammation in HFD-induced nonalcoholic steatohepatitis by STAT1β/TLR2 signaling pathway. Mol. Immunol. 2021, 134, 118–128.
- Wang, J.; Li, H.; Xue, B.; Deng, R.; Huang, X.; Xu, Y.; Chen, S.; Tian, R.; Wang, X.; Xun, Z.; et al. IRF1 Promotes the Innate Immune Response to Viral Infection by Enhancing the Activation of IRF3. J. Virol. 2020, 94, e01231-20.
- Zan, J.; Xu, R.; Tang, X.; Lu, M.; Xie, S.; Cai, J.; Huang, Z.; Zhang, J. RNA helicase DDX5 suppresses IFN-I antiviral innate immune response by interacting with PP2A-Cβ to deactivate IRF3. Exp. Cell Res. 2020, 396, 112332.
- Taneja, V.; Kalra, P.; Goel, M.; Khilnani, G.C.; Saini, V.; Prasad, G.; Gupta, U.D.; Krishna Prasad, H. Impact and prognosis of the expression of IFN-α among tuberculosis patients. PLoS ONE 2020, 15, e0235488.
- Antonczyk, A.; Krist, B.; Sajek, M.; Michalska, A.; Piaszyk-Borychowska, A.; Plens-Galaska, M.; Wesoly, J.; Bluyssen, H.A.R. Direct Inhibition of IRF-Dependent Transcriptional Regulatory Mechanisms Associated With Disease. Front. Immunol. 2019, 10, 1176.
- Miyamoto, M.; Fujita, T.; Kimura, Y.; Maruyama, M.; Harada, H.; Sudo, Y.; Miyata, T.; Taniguchi, T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell 1988, 54, 903–913.
- Yanai, H.; Negishi, H.; Taniguchi, T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Oncoimmunology 2012, 1, 1376–1386.
- Eguchi, J.; Yan, Q.W.; Schones, D.E.; Kamal, M.; Hsu, C.H.; Zhang, M.Q.; Crawford, G.E.; Rosen, E.D. Interferon regulatory factors are transcriptional regulators of adipogenesis. Cell Metab. 2008, 7, 86–94.
- Silvestre, M.F.; Kieswich, J.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C.; Caton, P.W. A key role for interferon regulatory factors in mediating early-life metabolic defects in male offspring of maternal protein restricted rats. Horm. Metab. Res. 2014, 46, 252–258.
- Eguchi, J.; Kong, X.; Tenta, M.; Wang, X.; Kang, S.; Rosen, E.D. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes 2013, 62, 3394–3403.
- Chen, M.; Wen, X.; Gao, Y.; Liu, B.; Zhong, C.; Nie, J.; Liang, H. IRF-4 deficiency reduces inflammation and kidney fibrosis after folic acid-induced acute kidney injury. Int. Immunopharmacol. 2021, 100, 108142.
- Li, Y.; Liu, Y.; Huang, Y.; Yang, K.; Xiao, T.; Xiong, J.; Wang, K.; Liu, C.; He, T.; Yu, Y.; et al. IRF-1 promotes renal fibrosis by downregulation of Klotho. FASEB J. 2020, 34, 4415–4429.
- Fabié, A.; Mai, L.T.; Dagenais-Lussier, X.; Hammami, A.; van Grevenynghe, J.; Stäger, S. IRF-5 Promotes Cell Death in CD4 T Cells during Chronic Infection. Cell Rep. 2018, 24, 1163–1175.
- Gapud, E.J.; Trejo-Zambrano, M.I.; Gomez-Banuelos, E.; Tiniakou, E.; Antiochos, B.; Granville, D.J.; Andrade, F.; Casciola-Rosen, L.; Rosen, A. Granzyme B Induces IRF-3 Phosphorylation through a Perforin-Independent Proteolysis-Dependent Signaling Cascade without Inducing Cell Death. J. Immunol. 2021, 206, 335–344.
- Sun, L.; Li, Y.; Misumi, I.; González-López, O.; Hensley, L.; Cullen, J.M.; McGivern, D.R.; Matsuda, M.; Suzuki, R.; Sen, G.C.; et al. IRF3-mediated pathogenicity in a murine model of human hepatitis A. PLoS Pathog. 2021, 17, e1009960.
- Liu, J.; Zhuang, Z.J.; Bian, D.X.; Ma, X.J.; Xun, Y.H.; Yang, W.J.; Luo, Y.; Liu, Y.L.; Jia, L.; Wang, Y.; et al. Toll-like receptor-4 signalling in the progression of non-alcoholic fatty liver disease induced by high-fat and high-fructose diet in mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 482–488.
- Shrivastava, S.; Meissner, E.G.; Funk, E.; Poonia, S.; Shokeen, V.; Thakur, A.; Poonia, B.; Sarin, S.K.; Trehanpati, N.; Kottilil, S. Elevated hepatic lipid and interferon stimulated gene expression in HCV GT3 patients relative to non-alcoholic steatohepatitis. Hepatol. Int. 2016, 10, 937–946.
- Klune, J.R.; Dhupar, R.; Kimura, S.; Ueki, S.; Cardinal, J.; Nakao, A.; Nace, G.; Evankovich, J.; Murase, N.; Tsung, A.; et al. Interferon regulatory factor-2 is protective against hepatic ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G666–G673.
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci. Immunol. 2017, 2, eaai7616.
- Roh, Y.S.; Kim, J.W.; Park, S.; Shon, C.; Kim, S.; Eo, S.K.; Kwon, J.K.; Lim, C.W.; Kim, B. Toll-Like Receptor-7 Signaling Promotes Nonalcoholic Steatohepatitis by Inhibiting Regulatory T Cells in Mice. Am. J. Pathol. 2018, 188, 2574–2588.
- Hart, K.M.; Fabre, T.; Sciurba, J.C.; Gieseck, R.L., 3rd; Borthwick, L.A.; Vannella, K.M.; Acciani, T.H.; de Queiroz Prado, R.; Thompson, R.W.; White, S.; et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-β. Sci. Transl. Med. 2017, 9.
- Imanishi, T.; Unno, M.; Kobayashi, W.; Yoneda, N.; Akira, S.; Saito, T. mTORC1 Signaling Controls TLR2-Mediated T-Cell Activation by Inducing TIRAP Expression. Cell Rep. 2020, 32, 107911.
- Yan, J.; Pandey, S.P.; Barnes, B.J.; Turner, J.R.; Abraham, C. T Cell-Intrinsic IRF5 Regulates T Cell Signaling, Migration, and Differentiation and Promotes Intestinal Inflammation. Cell Rep. 2020, 31, 107820.
- Aoki, Y.; Sugiyama, M.; Murata, K.; Yoshio, S.; Kurosaki, M.; Hashimoto, S.; Yatsuhashi, H.; Nomura, H.; Kang, J.H.; Takeda, T.; et al. Association of serum IFN-λ3 with inflammatory and fibrosis markers in patients with chronic hepatitis C virus infection. J. Gastroenterol. 2015, 50, 894–902.
- Petta, S.; Valenti, L.; Tuttolomondo, A.; Dongiovanni, P.; Pipitone, R.M.; Cammà, C.; Cabibi, D.; Di Marco, V.; Fracanzani, A.L.; Badiali, S.; et al. Interferon lambda 4 rs368234815 TT>δG variant is associated with liver damage in patients with nonalcoholic fatty liver disease. Hepatology 2017, 66, 1885–1893.
- Pelka, K.; Latz, E. IRF5, IRF8, and IRF7 in human pDCs-the good, the bad, and the insignificant? Eur. J. Immunol. 2013, 43, 1693–1697.
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924.
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135.
- Irving, A.T.; Zhang, Q.; Kong, P.S.; Luko, K.; Rozario, P.; Wen, M.; Zhu, F.; Zhou, P.; Ng, J.H.J.; Sobota, R.M.; et al. Interferon Regulatory Factors IRF1 and IRF7 Directly Regulate Gene Expression in Bats in Response to Viral Infection. Cell Rep. 2020, 33, 108345.
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325.
- Negishi, H.; Taniguchi, T.; Yanai, H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb. Perspect. Biol. 2018, 10, a028423.
- Liu, J.; Cao, S.; Herman, L.M.; Ma, X. Differential regulation of interleukin (IL)-12 p35 and p40 gene expression and interferon (IFN)-gamma-primed IL-12 production by IFN regulatory factor 1. J. Exp. Med. 2003, 198, 1265–1276.
- Zhang, C.; Yang, M.; Ericsson, A.C. Function of Macrophages in Disease: Current Understanding on Molecular Mechanisms. Front. Immunol. 2021, 12, 620510.
- Cui, H.; Banerjee, S.; Guo, S.; Xie, N.; Liu, G. IFN Regulatory Factor 2 Inhibits Expression of Glycolytic Genes and Lipopolysaccharide-Induced Proinflammatory Responses in Macrophages. J. Immunol. 2018, 200, 3218–3230.
- Fang, J.; Ji, Y.X.; Zhang, P.; Cheng, L.; Chen, Y.; Chen, J.; Su, Y.; Cheng, X.; Zhang, Y.; Li, T.; et al. Hepatic IRF2BP2 Mitigates Nonalcoholic Fatty Liver Disease by Directly Repressing the Transcription of ATF3. Hepatology 2020, 71, 1592–1608.
- Wang, X.A.; Zhang, R.; She, Z.G.; Zhang, X.F.; Jiang, D.S.; Wang, T.; Gao, L.; Deng, W.; Zhang, S.M.; Zhu, L.H.; et al. Interferon regulatory factor 3 constrains IKKβ/NF-κB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 2014, 59, 870–885.
- Benzler, J.; Ganjam, G.K.; Pretz, D.; Oelkrug, R.; Koch, C.E.; Legler, K.; Stöhr, S.; Culmsee, C.; Williams, L.M.; Tups, A. Central inhibition of IKKβ/NF-κB signaling attenuates high-fat diet-induced obesity and glucose intolerance. Diabetes 2015, 64, 2015–2027.
- Ni, Y.; Zhuge, F.; Nagashimada, M.; Ota, T. Novel Action of Carotenoids on Non-Alcoholic Fatty Liver Disease: Macrophage Polarization and Liver Homeostasis. Nutrients 2016, 8, 391.
- Huang, S.C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830.
- Vasanthakumar, A.; Moro, K.; Xin, A.; Liao, Y.; Gloury, R.; Kawamoto, S.; Fagarasan, S.; Mielke, L.A.; Afshar-Sterle, S.; Masters, S.L.; et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 2015, 16, 276–285.
- Fabrizi, M.; Marchetti, V.; Mavilio, M.; Marino, A.; Casagrande, V.; Cavalera, M.; Moreno-Navarrete, J.M.; Mezza, T.; Sorice, G.P.; Fiorentino, L.; et al. IL-21 is a major negative regulator of IRF4-dependent lipolysis affecting Tregs in adipose tissue and systemic insulin sensitivity. Diabetes 2014, 63, 2086–2096.
- Sindhu, S.; Kochumon, S.; Thomas, R.; Bennakhi, A.; Al-Mulla, F.; Ahmad, R. Enhanced Adipose Expression of Interferon Regulatory Factor (IRF)-5 Associates with the Signatures of Metabolic Inflammation in Diabetic Obese Patients. Cells 2020, 9, 730.
- Alzaid, F.; Lagadec, F.; Albuquerque, M.; Ballaire, R.; Orliaguet, L.; Hainault, I.; Blugeon, C.; Lemoine, S.; Lehuen, A.; Saliba, D.G.; et al. IRF5 governs liver macrophage activation that promotes hepatic fibrosis in mice and humans. JCI Insight 2016, 1, e88689.
- Tong, J.; Han, C.J.; Zhang, J.Z.; He, W.Z.; Zhao, G.J.; Cheng, X.; Zhang, L.; Deng, K.Q.; Liu, Y.; Fan, H.F.; et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology 2019, 69, 2471–2488.
- Wang, X.A.; Zhang, R.; Zhang, S.; Deng, S.; Jiang, D.; Zhong, J.; Yang, L.; Wang, T.; Hong, S.; Guo, S.; et al. Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E485–E495.
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068.
- Salem, S.; Salem, D.; Gros, P. Role of IRF8 in immune cells functions, protection against infections, and susceptibility to inflammatory diseases. Hum. Genet. 2020, 139, 707–721.
- Langlais, D.; Barreiro, L.B.; Gros, P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med. 2016, 213, 585–603.
- Berghout, J.; Langlais, D.; Radovanovic, I.; Tam, M.; MacMicking, J.D.; Stevenson, M.M.; Gros, P. Irf8-regulated genomic responses drive pathological inflammation during cerebral malaria. PLoS Pathog. 2013, 9, e1003491.
- Shiau, C.E.; Kaufman, Z.; Meireles, A.M.; Talbot, W.S. Differential requirement for irf8 in formation of embryonic and adult macrophages in zebrafish. PLoS ONE 2015, 10, e0117513.
- Yang, Q.; Yan, C.; Gong, Z. Interaction of hepatic stellate cells with neutrophils and macrophages in the liver following oncogenic kras activation in transgenic zebrafish. Sci. Rep. 2018, 8, 8495.
- Xu, H.; Zhu, J.; Smith, S.; Foldi, J.; Zhao, B.; Chung, A.Y.; Outtz, H.; Kitajewski, J.; Shi, C.; Weber, S.; et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol. 2012, 13, 642–650.
- Wang, X.A.; Zhang, R.; Jiang, D.; Deng, W.; Zhang, S.; Deng, S.; Zhong, J.; Wang, T.; Zhu, L.H.; Yang, L.; et al. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology 2013, 58, 603–616.
- Yang, Q.; Ding, W.; Cao, Y.; Zhou, Y.; Ni, S.; Shi, T.; Fu, W. Interferonregulatoryfactor-8(IRF-8) regulates the expression of matrix metalloproteinase-13 (MMP-13) in chondrocytes. Cell Stress Chaperones 2018, 23, 393–398.
- Chen, Y.J.; Liang, L.; Li, J.; Wu, H.; Dong, L.; Liu, T.T.; Shen, X.Z. IRF-2 Inhibits Gastric Cancer Invasion and Migration by Down-Regulating MMP-1. Dig. Dis. Sci. 2020, 65, 168–177.