Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Interferon regulatory factors (IRFs), a family of transcription factors that regulate IFN expression, play important roles in both innate and adaptive immune responses. IRF-involved signaling pathways contribute to hepatic inflammation, insulin resistance, and immune cell activation, such as macrophage polarization, playing critical roles in NAFLD or NASH pathogenesis. Treatments such as microRNAs, PPAR modulators, anti-inflammatory agents, and TLR agonists or antagonists that modulate IRF-mediated signaling pathways can ameliorate the progression of NAFLD to NASH.
- non-alcoholic fatty liver disease
- non-alcoholic steatohepatitis
- interferon
- interferon regulatory factor
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is becoming the most common chronic liver disease, accompanying the increased incidence of comorbidities, such as obesity, diabetes, and cardiovascular diseases. By 2030, NAFLD will affect more than 100 million people in the United States [1], posing a tremendous economic burden. The more severe form of NAFLD is non-alcoholic steatohepatitis (NASH) with the progression of liver inflammation and varying degrees of fibrosis, which can progress to cirrhosis and hepatocellular carcinoma (HCC) [2,3,4]. In addition, NAFLD may progress to HCC without the development of NASH or cirrhosis [5]. Factors such as sedentary lifestyle and overnutrition (consumption of high fructose) can cause NAFLD and its progression [6,7,8].
Interferons (IFNs) have been shown to play important roles in hepatic inflammation, fibrogenesis, and lipid accumulation. For example, feeding a methionine- and choline-deficient high-fat (MCDHF) diet induced more severe liver steatosis and inflammation on day 42 and advanced liver fibrosis on day 70 in IFN-γ-deficient, compared with wild-type mice [13]. In vitro study further showed that IFN-γ can stimulate the inflammatory response of macrophages and subsequent activation of hepatic stellate cells (HSCs), the collagen-producing cells in the liver, resulting in the progression of liver fibrosis [13]. Another study showed that IFN-γ was involved in the cytotoxicity of natural killer (NK) cells against HSCs in both mice and humans to attenuate liver fibrosis, which can be induced by activation of glutamate and metabotropic glutamate receptor 5 (mGluR5) in NK cells [14]. In addition, IFN-γ was upregulated in the NASH liver of rats, which promoted Toll-like receptor 2 (TLR2)-induced progression of liver inflammation [15].
Interferon regulatory factors (IRFs), a family of transcription factors that regulate IFN expression, play important roles in both innate and adaptive immune responses [16,17,18]. IRFs bind together with other transcriptional factors (e.g., signal transducer and activator of transcription/STAT, PU.1/Spi-1 proto-oncogene, and nuclear factor-kappa B/NF-κB) in the nucleus to regulate target gene expression [19]. IRF1 was initially found to regulate type I interferon secretion, including interferons alpha and beta (IFN-α and IFN-β) [20]. To date, it has been shown that there are nine members in the IRF family in humans and mice [21]. Compared with the white adipose tissue, liver tissue had higher mRNA expression levels of IRFs except for IRF4 in FVB mice [22]. IRFs have been shown to be involved in the regulation of adipogenesis and adipose tissue inflammation [23,24], renal fibrosis [25,26], and cell death [27,28]. Moreover, accumulating studies show that IRFs play important roles in chronic liver diseases [29,30,31,32], including NAFLD and NASH.
2. Interferons in NAFLD and NASH
Accumulating studies show that IFNs play pivotal roles in NAFLD and its progression to NASH, including type I, II, and III interferons. One study showed that the expression of type I interferon (IFN-I) was upregulated in human patients with NAFLD, which was positively associated with the number of CD8+ T cells [33]. Meanwhile, hepatic IFN-α protein level was found to increase in obese mice with activation of IRFs [33]. Another study showed that high levels of IFN-I induced apoptosis of regulatory T cells (Tregs), resulting in aggravation of NASH in mice [34]. Therefore, mice with depletion of IFN-α receptor 1 in CD8+ T cells were protected from the development of fatty liver disease [33]. In addition, NASH development was inhibited in IFN-α/β receptor 1-deficient mice than in wild-type mice [34].
IFN-γ is the only member of type II interferon. Hart et al. reported that IFN-γ-deficiency mice developed NASH more quickly, compared with wild-type mice on HFD, with the progression of transforming growth factor-β (TGF-β)-induced liver fibrosis [38]. IFN-γ was also upregulated in rat NASH liver, which was associated with TLR2-mediated liver inflammation [15]. TLR2/MyD88/NF-κB signaling pathway has been shown to be involved in T-cell activation and IFN-γ production [39]. Activation of IRF5 in CD4+ T cells can induce the production of Th1- and Th17-associated cytokines including IFN-γ, as well as chemokine receptors such as CCR5 (C-C chemokine receptor type 5) and CXCR4 (C-X-C motif chemokine receptor 4) but inhibit Th2-associated cytokines in mice [40].
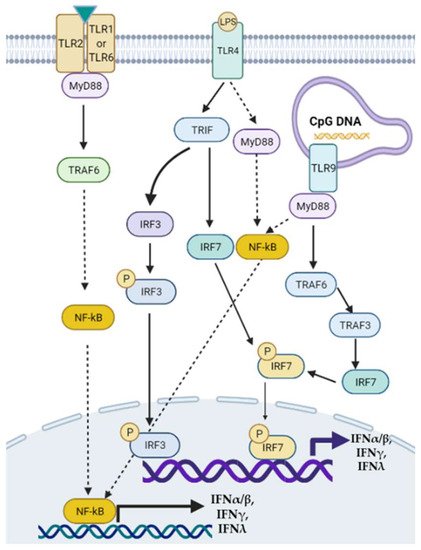
Type III interferons consist of four IFN-λ molecules—namely, IFN-λ1–4, known as antiviral cytokines. It has been demonstrated that serum levels of IFN-λ3 were increased in patients with chronic hepatitis C, compared with that in healthy controls, which was positively associated with the severity of liver fibrosis [42]. In addition, the rs368234815 TT allele on the IFN-λ4 locus was independently associated with advanced liver fibrosis, the severity of liver necroinflammation, and NASH [43]. CpG DNAs can stimulate TLR9 in murine pDCs via MyD88/TRAF6/TRAF3/IRF7/8 signaling pathway to regulate IFNα/β/λ secretion [44]. A graphic picture lists the signaling pathways of how TLR2, TLR4, TLR9, and IRFs regulate the production of IFNs (Figure 1).
Figure 1. Toll-like receptor-mediated signaling pathways of interferon (IFN) transcription. Modulation of TLRs such as TLR2, TLR4, and TLR9 can activate downstream MyD88/NF-κB, MyD88/TRAF6/3/IRF, or TRIF/IRF3/7 signaling pathways to regulate the transcription of type I, II, and III IFNs. TRAF: tumor necrosis factor receptor-associated factor.
Furthermore, IFN binding with IFN receptors can result in feedback regulation of IRF/IFN signaling in diseases [45,46,47]. Overall, IFNs are involved in the NAFLD/NASH pathogenesis, and their expression can be regulated by transcriptional factors including IRFs [48,49].
3. Roles of IRFs in NAFLD and NASH
The roles of IRFs in the pathogenesis of NAFLD and NASH are diverse. Herein, we discuss all nine members of IRF in liver inflammation, fibrogenesis, and cell death, which are closely associated with the progression of NAFLD to NASH.
3.1. IRF1
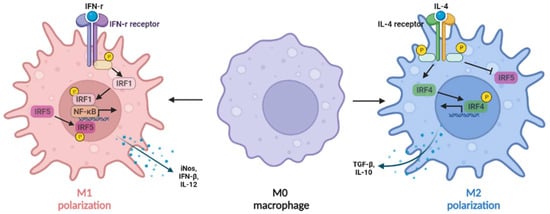
IFN-γ, which was highly expressed in rat NASH liver, induced the activation of transcriptional factors phosphorylated signal transducer and activator of transcription 1 (pSTAT1) and IRF1, resulting in upregulation of liver inflammation [15]. In addition, the expression of IRF1 in macrophages can be induced by IFN-γ (Figure 2), synergizing with NF-κB to induce M1-like macrophages with the production of IL-12, inducible nitric oxide synthase (iNOS), and IFN-β [50,51].
Figure 2. IRFs regulate macrophage polarization. The expression of IRF1 in macrophages can be induced by IFN-γ, synergizing with nuclear factor kappa B (NF-κB) to induce IL-12, inducible nitric oxide synthase (iNOS), and IFN-β production (M1-like macrophages), whereas IL-4 can induce M2-like macrophage polarization by activating IRF4 signaling to prevent IRF5-mediated M1 polarization.
3.2. IRF2
IRF2 shows an anti-inflammatory function in macrophages. Knockdown of IRF2 accelerated LPS-induced activation of macrophages by regulating hypoxia-inducible factor 1-alpha (HIF-1α)-dependent glycolysis [53]. In addition, IRF2 binding protein 2 (IRF2BP2) was significantly decreased in the fatty liver [54]. Specific knockdown of IRF2BP2 in hepatocytes aggravated HFD-induced hepatic steatosis, inflammation, and insulin resistance, indicating a protective role of IRF2BP2 in NAFLD. In contrast, overexpression of IRF2BP2 can inhibit these processes [54].
3.3. IRF3
Globally knocking out IRF3 dramatically promoted diet-induced hepatic steatosis and insulin resistance, whereas overexpression of IRF3 mediated by adenoviruses promoted a balance of energy metabolism, such as glucose and lipid [55]. In addition, the molecular study showed that the effect of IRF3 on hepatic steatosis was mediated by inhibition of the nuclear factor-kappa B kinase subunit beta (IKKβ)/NF-κB signaling pathway [55]. Inhibiting IKKβ/NF-κB signaling pathway has been shown to inhibit HFD-induced obesity and its comorbidities [56].
3.4. IRF4
In macrophages (Figure 2), IL-4 can induce M2 macrophage polarization by activating IRF4 signaling to prevent IRF5-mediated M1 macrophage polarization [59]. In addition, IRF4 is required for the mTORC2-mediated signaling pathway during M2 macrophage polarization [60]. In obesity, IRF4 is required for Treg differentiation except for basic leucine zipper transcription factor activating transcription factor-like (BATF), peroxisome proliferator-activated receptor gamma (PPAR-γ), and IL-33 [61]. IL-21 has been reported to be a negative regulator of IRF4, and IL-21-deficient mice decreased fat accumulation and liver inflammation [62], indicating a protective role of IRF4 in NAFLD. In vitro study also showed that IRF4 plays an important role in IL-4 induced expression of extracellular matrix proteins, such as α-smooth muscle actin (α-SMA), as well as the transition of M2-like macrophages from mouse bone marrow-derived monocytes to myofibroblasts [25].
3.5. IRF5
The expression of IRF5 was increased in adipose tissue associated with metabolic syndrome in patients with obesity or type 2 diabetes [63]. In addition, the expression of IRF5 was positively correlated with inflammatory markers (e.g., TNF-α and IL-23A), which may function as a marker of metabolic inflammation. Alzaid et al. reported that IRF5 was upregulated in hepatic macrophages from human patients with liver fibrosis induced by NAFLD or hepatitis C virus infection [64]. Mice with IRF5 knockdown in myeloid cells were protected from metabolic stress or toxin-induced liver fibrosis, compared with wild-type controls [64]. In addition, IRF5 expression was positively associated with proinflammatory cytokines such as TNF, IL-1β, and IL-6, and was negatively associated with anti-inflammatory cytokines such as IL-10 and profibrotic gene TGF-β1.
3.6. IRF6
IRF6 was significantly decreased in HFD-induced fatty liver. A cellular mechanism study showed that knockout IRF6 specifically in hepatocytes accelerated liver steatosis, while overexpression of IRF6 in hepatocytes ameliorated liver steatosis [67]. A molecular mechanism study exhibited that IRF6 can directly bind to the promoter of the peroxisome proliferator-activated receptor γ (PPARγ) gene, resulting in suppression of PPARγ expression and its targeted genes to inhibit lipid accumulation [67].
3.7. IRF7
The expression of IRF7 was upregulated in liver tissue of diet-induced obese mice and genetically obese (ob/ob) mice, compared with that in lean controls [68]. IRF7-deficient mice displayed less body weight, insulin sensitivity, hepatic macrophage infiltration, inflammation, and steatosis, compared with wild-type controls [68]. A complex formed by MyD88, TRAF6, and IRF7 is required for TLR-mediated IFN-α expression, and IRF7 activation requires the ubiquitin E3 ligase activity of TRAF6 [69].
3.8. IRF8
Together with IRF1, IRF8 plays an essential role in IFN-γ induced activation of macrophages, as well as the function of myeloid cells [70]. Loss of IRF8 may cause more severe infection, as it regulates the expression of antimicrobial and inflammatory genes in mice with neuroinflammation or pulmonary tuberculosis [71,72]. Knocking down IRF8 in zebrafish caused a reduction in macrophage numbers and the number and activation of hepatic stellate cells [73,74]. The Notch/RBPJ (recombination signal binding protein for immunoglobulin kappa J region) signaling can control the expression of IRF8 to modulate M1 macrophage cytokine expression [75].
3.9. IRF9
Interestingly, Wang et al. reported that IRF9 displayed a contrary role in obese mice compared to IRF7. The hepatic expression of IRF9 was decreased in the liver tissues of obese mice, compared with that in lean controls [76]. IRF9 knockout (KO) mice showed increased insulin resistance, hepatic steatosis, and inflammation compared to wild-type controls while feeding HFD. In contrast, overexpression of IRF9 mediated by adenoviruses significantly improved the metabolic syndrome in HFD-fed mice or ob/ob obese mice [76].
Overall, IRFs show diverse functions in fatty liver, liver fibrosis, and NAFLD (Table 1), including that IRF2, 3, 4, 6, and 9 show a protective role in NAFLD, whereas IRF1, 5, 7, and 8 exhibit an accelerated function on NAFLD. In addition, IRFs have been shown to regulate the expression of matrix metalloproteinases (MMPs) in other tissues or organs that play an important role in matrix destruction or degradation [77,78]. Noticeably, the same IRF may function variably in different liver resident cells. Selectively regulating IRF expression may provide therapeutic options for NAFLD and NASH.
Table 1. The role of IRFs in NAFLD and NASH.
| IRFs | Model | Expression * | Function | References |
|---|---|---|---|---|
| IRF1 | NASH rat | Increased | IFN-γ in rat NASH liver upregulated IRF1 expression, resulting in liver inflammation progression. | [15] |
| IRF2 | M1-like macrophages | Decreased | Knockdown of IRF2 accelerated lipopolysaccharide (LPS)-induced activation of macrophages by regulating hypoxia-inducible factor 1-alpha (HIF-1α)-dependent glycolysis. | [53] |
| IRF3 | NAFLD mice | Decreased | IRF3 deficiency dramatically promoted diet-induced hepatic steatosis, and insulin resistance, whereas overexpression of IRF3 induced hemostasis of glucose and lipid balance metabolism, via regulating nuclear factor-kappa B kinase subunit beta (IKKβ)/nuclear factor kappa B (NF-κB) signaling pathway. | [55] |
| IRF4 | M2-like macrophages | Increased | IRF4 is involved in M2-like macrophage polarization induced by IL-4 or mediated by the mTORC2 signaling pathway. | [59,60] |
| IRF5 | Mice with liver fibrosis | Increased | Mice with IRF5 knockdown in myeloid cells were protected from metabolic stress or toxin-induced liver fibrosis, compared with wild-type controls. | [64] |
| IRF6 | NAFLD mice | Decreased | Cellular mechanism study showed that knockout IRF6 specifically in hepatocytes accelerated liver steatosis, while overexpression of IRF6 in hepatocytes ameliorated liver steatosis. | [67] |
| IRF7 | Obese mice | Increased | IRF7 deficiency reduced body weight, insulin resistance, hepatic macrophage infiltration, inflammation, and steatosis in mice on a high-fat diet (HFD). | [68] |
| IRF8 | Zebrafish with liver fibrosis | Increased | Knocking down IRF8 in zebrafish caused a reduction in macrophage numbers and the number and activation of hepatic stellate cells. | [73,74] |
| IRF9 | Obese mice | Decreased | IRF9 knockout increased insulin resistance, hepatic steatosis, inflammation in mice on HFD. | [76] |
* The increase or decrease in IRF denotes its expression during the progression of NAFLD, liver fibrosis, and macrophage polarization. The Table is from Zhang, C.; Liu, S.; Yang, M. The Role of Interferon Regulatory Factors in Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Gastroenterol. Insights 2022, 13, 148-161. https://doi.org/10.3390/gastroent13020016.
This entry is adapted from the peer-reviewed paper 10.3390/gastroent13020016
This entry is offline, you can click here to edit this entry!