Vitamin D was found to counteract insulin resistance via participation in the maintenance of normal resting reactive oxygen species level and regulation of Ca2+ level in many cell types. Both genomic and non-genomic action of vitamin D is directed to insulin signaling. Thereby, vitamin D reduces the extent of pathologies associated with insulin resistance such as oxidative stress and inflammation. Therefore, the beneficial actions of vitamin D include an improvement of glucose and lipid metabolism in insulin-sensitive tissues, and in consequence the diminish of insulin resistance.
- vitamin D, insulin resistance,
- inflammation
- oxidative stress
1. Introduction
There is mounting evidence that vitamin D deficiency is now a worldwide health problem. In addition, an alarming number of diseases connected with vitamin D deficiency such as obesity and type 2 diabetes mellitus (T2DM) are observed. Both basic and clinical studies demonstrated that the majority of common characteristics of these diseases result from defects in insulin signaling, systemic inflammation, and pancreatic β-cells dysfunction [1][2][3][4]. It should be stressed that according to recent investigations one of the major causative factors in insulin resistance development is vitamin D deficiency. The results of some clinical studies have demonstrated that vitamin D supplementation improves major metabolic parameters associated with insulin resistance, including low-density lipoprotein (LDL), total cholesterol (TC), glycated hemoglobin (HbA1c), triglyceride (TAG), and homeostatic model assessment-insulin resistance (HOMA-IR). We have shown that three-month supplementation with vitamin D of the elderly with metabolic disorders markedly elevates HDL level, reduces HOMA-IR, and TG/HDL ratio. Moreover, we observed that HbA1c percentage decreased about 0.5% in T2DM patients after vitamin D supplementation [5]. In turn, Upreti et al. have revealed that six-month supplementation with vitamin D of T2DM patients leads to distinct reduction of HbA1c [6]. The results of study carried out by Mirrhosseini et al. have showed that vitamin D decreases HbA1c, fasting plasma glucose (FPG), and HOMA-IR contributing to better glycemic control [7]. Interestingly, Tabesh et al. have observed that co-supplementation of vitamin D with calcium decreases serum insulin level, HbA1c, HOMA-IR, LDL, and TC/HDL. Additionally, they also detected the significant elevation of quantitative insulin sensitivity check index (QUICKI) and HDL [8]. El Hajj et al. have found that vitamin D triggers to significantly diminish of HOMA-IR, FPG, TC, and LDL, but without any significant changes in HbA1c [9]. The results of studies conducted by Barzegardi et al. have presented pronounced decrease in serum levels of TG, LDL, and TC in diabetic nephropathy patients after supplementation with vitamin D [10]. Taken together, these observations support that vitamin D improves metabolic control of diabetes.
Vitamin D is involved in many cellular processes, e.g., the presence of its receptor and its metabolizing enzymes have been found in the cells of various tissues, including pancreatic β-cells, adipocytes, hepatocytes, and myocytes [11][12][13][14]. It also controls blood glucose concentration by regulating insulin secretion and insulin sensitivity [15]. Furthermore, it has been found to act in adipose tissue which is a major storage site of the vitamin [11]. It should be underlined that adipose tissue secretes numerous adipocytokines involved in inflammation, a typical feature of insulin resistance, obesity, and T2DM [11]. Numerous studies have revealed that vitamin D reduces the extent of inflammation and chronic hyperglycemia-generated oxidative stress [5][15]. Appealingly, vitamin D was demonstrated to modulate hepatic lipid and glucose metabolism [16]. Finally, it has also been shown that vitamin D counteracts diet-induced insulin resistance in skeletal muscle [17].
However, it should be also emphasized that the results of clinical studies have revealed no effect of vitamin D on insulin resistance and related disorders, including oxidative stress and inflammation. Lerchbaum et al. have shown that vitamin D supplementation did not change significantly metabolic parameters regarding insulin resistance and lipids in heathy men [18]. Forouhi et al. have found no effect of vitamin D on HbA1c, lipid and apolipoprotein levels, CRP, as well as anthropometric measures in subjects with increased risk of T2DM [19]. Similarly, Heshmat et al. have revealed no changes in HbA1c, anthropometric measures, and HOMA-IR in diabetic patients treated with vitamin D [20]. No differences in the FPG oral glucose tolerance test (OGTT) between prediabetes subjects supplemented with vitamin D in comparison to the placebo group have also been observed [21]. In addition, no significant changes between T2DM group and T2DM group supplemented with vitamin D have also been observed in the hs-CRP level, oxidative stress markers, LDL, HDL, and HbA1c [22]. In turn, Asemi et al. did not observe any significant changes in total plasma glutathione (GSH) and serum high sensitivity C-reactive protein (hs-CRP) level in pregnant women with gestational diabetes after supplementation with vitamin D [23].
2. How does Vitamin D Overcome Insulin Resistance and Related Disorders?
2.1. Vitamin D via the Regulation of Ca2+ Homeostasis Participates in Insulin Secretion by Pancreatic β-Cells
The secretion of insulin by the pancreatic β-cells is a consequence of elevated blood glucose concentration. Glucose molecules flow into the pancreatic β-cells via the glucose transporter 2 (GLUT-2). Then, glucose breaks down in numerous metabolic pathways, which is ultimately accompanied by ATP production. Increased ATP suppresses the ATP-sensitive K+ channel resulting in the depolarization of β-cell membrane followed by the activation of the L-type voltage-operated channels to produce the localized Ca2+ pulses crucial for the secretion of insulin [24].
Numerous studies showed that vitamin D deficiency is associated with impaired secretion of insulin by pancreatic β-cells [25][26][27][28][29]. Importantly, it was demonstrated that the supplementation with vitamin D restored proper secretion of the hormone [25][27][30]. However, it should be underlined that the findings concerning this issue are not unambiguous especially with regard to clinical trials [3][31][32][33][34][35].
One of the molecular mechanisms by which vitamin D participates in insulin secretion by pancreatic β-cells is the regulation of intracellular Ca2+ concentration. It was reported that 1,25(OH)2D3 reduced the expression of the L-type Ca2+ channels causing a decrease in intracellular Ca2+ concentration and thereby altering calcium signaling. In turn, rapid, non-genomic 1,25(OH)2D3 action was found to be responsible for the increase of cytoplasmic Ca2+ level that activates exocytosis of insulin in the pancreatic β-cells. Two vitamin D-mediated signaling pathways are involved in this process. The first includes PKA activation that phosphorylates various proteins engaged in the function of L-type voltage-dependent Ca2+ channels associated with insulin secretion. The second engages PLC synthesis and the activation of inositol triphosphate (InsP3) triggering the secretion of Ca2+ from ER leading to DAG synthesis. Subsequently, DAG activates PKC that is responsible for the phosphorylation of the KATP channels and L-type voltage-dependent Ca2+ channels. The latter trigger the depolarization of cytoplasmic membrane and opening of T-type Ca2+ and L-type channels that in consequence leads to the elevation of intracellular Ca2+ followed by insulin secretion [36]. PKC is also able to mobilize insulin secretory vesicles that together with increased Ca2+ concentration induce insulin secretion [37]. Furthermore, increased intracellular Ca2+ concentration stimulates insulin secretion via activation of CaMPKII. CaMPKII is a serine-threonine protein kinase occurring in secretory vesicles of insulin. Its primary function is the promotion of phosphorylation of numerous proteins involved in exocytosis, as well as mobilization of insulin vesicles [38]. Another study demonstrated that increased intracellular Ca2+ concentration is associated with the expression of insulin gene via cAMP-responsive element-binding protein (CREB). The activation of CREB occurs in response to numerous stimuli, including glucose growth factors (i.e., the insulin-like growth factor-1 (IGF-1)), incretin hormones (i.e., the glucagon-like peptide-1 (GLP-1), the gastric inhibitory polypeptide (GIP), the pituitary adenylate cyclase-activating polypeptide (PACAP)). All these stimuli lead to the phosphorylation of CREB at serine 133 residue. CREB is a crucial transcriptional element responsible for the efficient transcription of insulin gene, glucose sensing, exocytosis of insulin, and survival of pancreatic β-cells [39].
It is worth highlighting that the regulation of intracellular Ca2+ level by vitamin D is mediated by calbidin, a cytosolic Ca2+ -binding protein involved in the stimulation of insulin secretion. Calbidin-D28k expression was found to be regulated by vitamin D [33][40]. It was also reported that 1,25(OH)2D3 increased the expression of calbindin D-9k, parvalbumin, the plasma membrane Ca2+-ATPase 1b, the sodium/calcium exchanger (NCX), and the Ca2+ pumps. All of these proteins are involved in the maintenance of low resting Ca2+ concentration in pancreatic β-cells [41][42][43][44].
Taken together, vitamin D is a potential modulator of depolarization-induced secretion of insulin via intracellular Ca2+ level regulation in pancreatic β-cells [40]. The effect of vitamin D on pancreatic β-cells is presented in Figure 1.
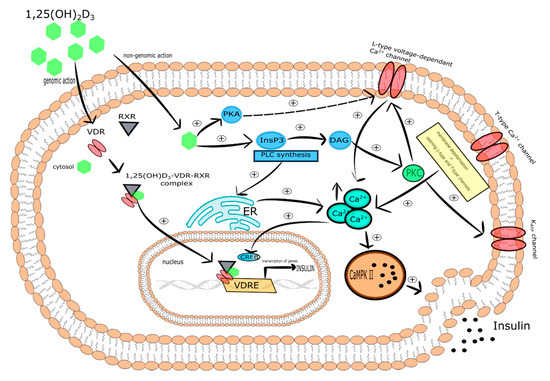
Figure 1. The effect of vitamin D on pancreatic β-cells. Stimulatory interactions are indicated by solid arrows and attenuation by dotted arrows. Enhancement is expressed by +. ↑ denotes increase.
2.2. Vitamin D Controls Ca2+ Level in Myocytes and Adipocytes
It is well known that Ca2+ are second messengers engaged in intracellular events induced by insulin in muscle and adipose tissue. That is why intracellular Ca2+ level changes have a substantial impact on multidirectional insulin actions. Numerous studies have demonstrated that the low level of Ca2+ in the cells of insulin targeted tissues is associated with reduced activity of glucose transporter followed by the development of peripheral insulin resistance [30].
Intracellular Ca2+ concentration in insulin-responsive tissues, including adipose tissue and skeletal muscle, is regulated by several mechanisms. The first mechanism involves the action of PTH that increases intracellular Ca2+ concentration in insulin-responsive tissues, including adipose tissue and skeletal muscle [45][46][47], as well as reducing insulin-induced transport of glucose [48][49]. Therefore, both growing intracellular Ca2+ concentration and the decreasing number of GLUT-1 and GLUT-4 on the cell membranes evoked by PTH promotes insulin resistance observed as reduced glucose uptake [49][50]. There is evidence that vitamin D deficiency is associated with increased PTH levels coexisting with insulin resistance [51][52]. Wright et al. have shown that vitamin D reduced insulin resistance in skeletal muscle as a result of elevation of intracellular Ca2+ concentration and strengthening of GLUT-4 translocation to the membrane of muscle cells and glucose uptake [53].
It has also been observed that vitamin D might decrease insulin resistance indirectly via the renin-angiotensin-aldosterone system (RAAS). It is well known that the RAAS system inhibits insulin action in peripheral tissues and regulates cellular Ca2+ concentration in skeletal muscle cells [53][54][55]. Interestingly, the increased expression of renin and secretion of angiotensin II, as well as 1,25(OH)2D3-mediated inhibition of renin biosynthesis have been observed in VDR-null mice [56][57][58]. Therefore, it was shown that vitamin D improved insulin sensitivity via inhibition of RAAS [59].
To conclude, vitamin D alleviates the insulin resistance state via regulation of Ca2+ level and RAAS action in insulin targeted tissue, including skeletal muscle and adipose tissue.
2.3. Vitamin D-Mediated Improvement of Insulin Sensitivity Is Connected with Insulin Signaling
Accumulating evidence uncovers multiple potential mechanisms by which vitamin D deficiency can contribute to insulin resistance. It is generally accepted that abnormalities in the insulin signaling pathway are responsible for the development of insulin resistance that is characterized by reduced reaction of target cells to circulating insulin.
It has been found that vitamin D mediated increase in insulin sensitivity occurs via binding of calcitriol to VDR [60], induction of IRs expression [61], and the activation of peroxisome proliferator-activated receptor delta (PPAR-δ) [62]. The latter is a transcription factor engaged in the mobilization and metabolism of fatty acids in skeletal muscle and adipose tissue. What is more, activated PPAR-δ decreased FFAs-mediated insulin resistance in skeletal muscle. It was shown that 1,25(OH)2D3 activated PPAR-δ and improved insulin sensitivity. [62]. Manna et al. documented that vitamin D improved glucose metabolism as a result of upregulation of the SIRT1/IRS1/GLUT-4 signaling cascade and enhanced glucose uptake in high glucose-treated C2C12 myotubes [63].
The genomic pathway induced by 1,25(OH)2D3 in pancreatic β-cells, which express both VDR and CYP27B1, stimulates insulin synthesis and secretion since VDRE is present in the promoter region of the insulin gene [33][36][64]. Relevantly, studies performed on mice with the lack of functional VDR revealed that after glucose load, insulin synthesis and secretion were impaired [65]. Vitamin D-mediated improvement of insulin sensitivity is connected with insulin signaling. As a result of 1,25(OH)2D3 -mediated transcriptional activation of IR gene, the number of IRs on the surface of insulin responsive cells increases. Thus, upregulation of the IR gene ensures proper insulin signaling [61][66][67] and in this way calcitriol maintains insulin sensitivity [61][66][68]. It is suggested that vitamin D deficiency is involved in the onset of insulin resistance as a consequence of reduced expression of IR [2]. However, the results of vitamin D-mediated activation of IR expression in the liver are unambiguous. George et al. reported that vitamin D supplementation upregulated liver expression of IRs in streptozotocin-induced diabetic rats [69]. On the contrary, several studies failed to identify alterations in IR expression in the liver of mice fed with high-fat diet or low-fat diet [70], as well as in streptozotocin-induced diabetic rats after vitamin D supplementation [71].
To sum up, vitamin D alleviates insulin resistance via improvement of insulin signaling.
2.4. Vitamin D Possesses Indirect Antioxidant Properties
The pathogenic mechanism of insulin resistance is complex and has yet to be fully elucidated. Undoubtedly, the trigger factor in insulin resistance is adiposity, especially visceral, which is accompanied by chronic hyperglycemia, oxidative stress, and low grade chronic inflammation [72][73]. Additionally, a balance in the physiologic redox state is crucial for normal β-cells function, glucose homeostasis, and insulin sensitivity [74][75][76]. Oxidative stress is an imbalance between the production of reactive oxygen species (ROS) and the efficacy of antioxidant defense system. Endoplastic reticulum (ER) stress, hyperglycemia, dyslipidemia, lipid peroxides, and nitric oxide synthase, as well as advanced glycation end-products are involved in ROS overproduction in the insulin resistance diabetic state. It is well recognized that oxidative stress may activate several factors contributing to the development of insulin resistance [77][78]. Inoguchi et al. found that hyperglycemia and FFAs might activate ROS production via PKC-dependent stimulation of NADPH oxidase [79]. It was also observed that increased production of ROS is a key activator of insulin resistance [80][81]. Moreover, the association between the degree of insulin resistance and oxidative stress is suggested to induce cellular damage [78][82][83]. ROS have the ability to directly oxidize and damage cellular macromolecules, i.e., DNA, proteins, and lipids. Additionally, ROS may act as a signaling molecule that activates numerous cellular stress-sensitive pathways, i.e., NF-κB, JNK/SAPK, p38MAPK, and hexosamine involved in cellular damage and related pancreatic β-cells dysfunction, insulin resistance, and diabetes complication [84].
It is generally known that the hyperglycemic state is a causative factor responsible for the overproduction of ROS and reduced ATP formation that in turn exerts an effect on Ca2+ homeostasis leading to β-cell exhaustion and reduced resting insulin secretion. Furthermore, it is well recognized that the elevated formation of ROS increases the release of Ca2+ from ER via sensitization of the ryanodine receptors (RYRs) and inositol 1,4,5-trophosphate receptors (InsP3Rs). Reduced ATP level diminishes the capability of the Ca2+ pumps in ER and plasma membrane to press out Ca2+ from the cytoplasm outside of the cell. The effect may stimulate an increase of Ca2+ level in the pancreatic β-cells that triggers excessive insulin secretion leading to exhaustion of pancreatic β-cells [2]. Therefore, the elevated level of ROS strengthens Ca2+ signaling and may contribute to the onset of diabetes.
It was also proposed that oxidative stress coexisting with diabetes/chronic hyperglycemia is a result of increased FFAs levels that exert an effect on the mitochondria leading to increased ROS production (i.e., superoxide, hydrogen peroxide, hydroxyl radical ions) [85][86][87][88]. It was also suggested that vitamin D may regulate cellular bioenergetics in the mitochondria via VDR in the nucleus. This effect is related to the upregulation of numerous components involved in mitochondrial function, including mitochondrial respiration [89][90]. Additionally, VDR is capable of entering mitochondrion via permeability transition pores [91] and controls its functions, however this mechanism is still not fully understood [92]. It has also been found that vitamin D deficiency is connected with a decline in the mitochondrial respiration process. This effect is a consequence of the reduction of proteins and nuclear mRNA molecules engaged in this process [89][90]. Decreased respiration leads to a drop of mitochondrial bioenergetics related to alterations in oxidative phosphorylation, reduced ATP formation, and increased production of ROS [2]. Reduced expression of complex 1 of the electron transport chain contributes to the decrease of ATP production and ROS overproduction. In turn, increased ROS level reduces the activity of the insulin signaling pathways via lowering of GLUT-4 gene transcription, phosphorylation of IRS, disturbances in insulin signaling, and changes of mitochondrial activity [93][94][95]. These observations are supported by the results of a study showing that 1,25(OH)2D3/VDR signaling inhibits the process of differentiation of brown adipose cells and mitochondrial respiration [96]. Recently, Ricca et al. have demonstrated that VDR-mediated action of vitamin D may protect cells from overproduction of ROS and excessive respiration that leads to cell damage [97]. Vitamin D controls the balance of mitochondrial respiration via maintenance of mitochondrial respiratory chain activity [98] and the regulation of expression of uncoupling protein 1 (UCP1). UCP1 is localized on the inner membrane of mitochondria and is engaged in the regulation of thermogenesis [11]. The role of vitamin D in the maintenance of normal activity of mitochondria may explain at least partially the privileged relationship between diabetes and vitamin D deficiency.
Vitamin D has been shown to decrease ROS production in adipocytes [99] via the regulation of cellular antioxidants expression such as glucose-6-phosphate dehydrogenase (G6PD), glutathione peroxidase (Gpx), TR [100]. Interestingly, vitamin D together with Klotho and Nrf2 may control the expression of numerous antioxidants including catalase, Prx-2, Prx-3, SOD ½, GSH, TR, G6PD, TRX, Trxrd-1, Gpx. It has been documented that vitamin D decreases the expression of NADPH oxidase which is responsible for the production of ROS [101], while increasing the expression of SOD [100][102]. Furthermore, vitamin D elevated the production of glutathione (GSH), a major redox buffer through the upregulation of glutamate cysteine ligase, glutathione reductase, and G6PD [103][104][105]. To conclude, it seems that antioxidant properties of vitamin D are indirect and related to its genomic and non-genomic action.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21186644
References
- Wang, H.; Chen, W.; Li, D.; Yin, X.; Zhang, X.; Olsen, N.; Zheng, S.G. Vitamin D and Chronic Diseases. Aging Dis. 2017, 8, 346–353, doi:10.14336/AD.2016.1021.
- Berridge, M.J. Vitamin D deficiency and diabetes. Biochem. J. 2017, 474, 1321–1332, doi:10.1042/BCJ20170042.
- Al-Shoumer, K.A.; Al-Essa, T.M. Is there a relationship between vitamin D with insulin resistance and diabetes mellitus? World J. Diabetes 2015, 6, 1057–1064, doi:10.4239/wjd.v6.i8.1057.
- Tao, S.; Yuan, Q.; Mao, L.; Chen, F.-L.; Ji, F.; Cui, Z.-H. Vitamin D deficiency causes insulin resistance by provoking oxidative stress in hepatocytes. Oncotarget 2017, 8, 67605–67613, doi:10.18632/oncotarget.18754.
- Wenclewska, S.; Szymczak-Pajor, I.; Drzewoski, J.; Bunk, M.; Śliwińska, A. Vitamin D Supplementation Reduces Both Oxidative DNA Damage and Insulin Resistance in the Elderly with Metabolic Disorders. Int. J. Mol. Sci. 2019, 20, 2891, doi:10.3390/ijms20122891.
- Upreti, V.; Maitri, V.; Dhull, P.; Handa, A.; Prakash, M.S.; Behl, A. Effect of oral vitamin D supplementation on glycemic control in patients with type 2 diabetes mellitus with coexisting hypovitaminosis D: A parellel group placebo controlled randomized controlled pilot study. Diabetes Metab. Syndr. 2018, 12, 509–512, doi:10.1016/j.dsx.2018.03.008.
- Mirhosseini, N.; Vatanparast, H.; Mazidi, M.; Kimball, S.M. The Effect of Improved Serum 25-Hydroxyvitamin D Status on Glycemic Control in Diabetic Patients: A Meta-Analysis. J. Clin. Endocrinol. Metab. 2017, 102, 3097–3110, doi:10.1210/jc.2017-01024.
- Tabesh, M.; Azadbakht, L.; Faghihimani, E.; Tabesh, M.; Esmaillzadeh, A. Effects of calcium-vitamin D co-supplementation on metabolic profiles in vitamin D insufficient people with type 2 diabetes: A randomised controlled clinical trial. Diabetologia 2014, 57, 2038–2047, doi:10.1007/s00125-014-3313-x.
- El Hajj, C.; Chardigny, J.-M.; Boirie, Y.; Yammine, K.; Helou, M.; Walrand, S. Effect of Vitamin D Treatment on Glucose Homeostasis and Metabolism in Lebanese Older Adults: A Randomized Controlled Trial. J. Nutr. Health Aging 2018, 22, 1128–1132, doi:10.1007/s12603-018-1083-8.
- Barzegari, M.; Sarbakhsh, P.; Mobasseri, M.; Noshad, H.; Esfandiari, A.; Khodadadi, B.; Gargari, B.P. The effects of vitamin D supplementation on lipid profiles and oxidative indices among diabetic nephropathy patients with marginal vitamin D status. Diabetes Metab. Syndr. 2019, 13, 542–547, doi:10.1016/j.dsx.2018.11.008.
- Abbas, M.A. Physiological functions of Vitamin D in adipose tissue. J. Steroid. Biochem. Mol. Biol. 2017, 165, 369–381, doi:10.1016/j.jsbmb.2016.08.004.
- Ceglia, L. Vitamin D and Its Role in Skeletal Muscle. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 628–633, doi:10.1097/MCO.0b013e328331c707.
- Bischoff, H.A.; Borchers, M.; Gudat, F.; Duermueller, U.; Theiler, R.; Stähelin, H.B.; Dick, W. In situ detection of 1,25-dihydroxyvitamin D3 receptor in human skeletal muscle tissue. Histochem. J. 2001, 33, 19–24, doi:10.1023/a:1017535728844.
- Ding, N.; Liddle, C.; Evans, R.M.; Downes, M. Hepatic actions of Vitamin D receptor ligands: An unexpected solution to chronic liver disease? Expert Rev. Clin. Pharmacol. 2013, 6, 597–599, doi:10.1586/17512433.2013.841078.
- Wimalawansa, S.J. Associations of vitamin D with insulin resistance, obesity, type 2 diabetes, and metabolic syndrome. J. Steroid. Biochem. Mol. Biol. 2018, 175, 177–189, doi:10.1016/j.jsbmb.2016.09.017.
- Leung, P.S. The Potential Protective Action of Vitamin D in Hepatic Insulin Resistance and Pancreatic Islet Dysfunction in Type 2 Diabetes Mellitus. Nutrients 2016, 8, 147, doi:10.3390/nu8030147.
- Benetti, E.; Mastrocola, R.; Chiazza, F.; Nigro, D.; D’Antona, G.; Bordano, V.; Fantozzi, R.; Aragno, M.; Collino, M.; Minetto, M.A. Effects of vitamin D on insulin resistance and myosteatosis in diet-induced obese mice. PLoS ONE 2018, 13, e0189707, doi:10.1371/journal.pone.0189707.
- Lerchbaum, E.; Trummer, C.; Theiler-Schwetz, V.; Kollmann, M.; Wölfler, M.; Pilz, S.; Obermayer-Pietsch, B. Effects of Vitamin D Supplementation on Body Composition and Metabolic Risk Factors in Men: A Randomized Controlled Trial. Nutrients 2019, 11, 1894, doi:10.3390/nu11081894.
- Forouhi, N.G.; Menon, R.K.; Sharp, S.J.; Mannan, N.; Timms, P.M.; Martineau, A.R.; Rickard, A.P.; Boucher, B.J.; Chowdhury, T.A.; Griffiths, C.J.; et al. Effects of vitamin D2 or D3 supplementation on glycaemic control and cardiometabolic risk among people at risk of type 2 diabetes: Results of a randomized double-blind placebo-controlled trial. Diabetes Obes. Metab. 2016, 18, 392–400, doi:10.1111/dom.12625.
- Heshmat, R.; Tabatabaei-Malazy, O.; Abbaszadeh-Ahranjani, S.; Shahbazi, S.; Khooshehchin, G.; Bandarian, F.; Larijani, B. Effect of vitamin D on insulin resistance and anthropometric parameters in Type 2 diabetes; a randomized double-blind clinical trial. DARU J. Pharm. Sci. 2012, 20, 10, doi:10.1186/2008-2231-20-10.
- Davidson, M.B.; Duran, P.; Lee, M.L.; Friedman, T.C. High-dose vitamin D supplementation in people with prediabetes and hypovitaminosis D. Diabetes Care 2013, 36, 260–266, doi:10.2337/dc12-1204.
- Yiu, Y.-F.; Yiu, K.-H.; Siu, C.-W.; Chan, Y.-H.; Li, S.-W.; Wong, L.-Y.; Lee, S.W.L.; Tam, S.; Wong, E.W.K.; Lau, C.-P.; et al. Randomized controlled trial of vitamin D supplement on endothelial function in patients with type 2 diabetes. Atherosclerosis 2013, 227, 140–146, doi:10.1016/j.atherosclerosis.2012.12.013.
- Asemi, Z.; Hashemi, T.; Karamali, M.; Samimi, M.; Esmaillzadeh, A. Effects of vitamin D supplementation on glucose metabolism, lipid concentrations, inflammation, and oxidative stress in gestational diabetes: A double-blind randomized controlled clinical trial. Am. J. Clin. Nutr. 2013, 98, 1425–1432, doi:10.3945/ajcn.113.072785.
- Gilon, P.; Chae, H.-Y.; Rutter, G.A.; Ravier, M.A. Calcium signaling in pancreatic β-cells in health and in Type 2 diabetes. Cell Calcium 2014, 56, 340–361, doi:10.1016/j.ceca.2014.09.001.
- Cade, C.; Norman, A.W. Vitamin D3 improves impaired glucose tolerance and insulin secretion in the vitamin D-deficient rat in vivo. Endocrinology 1986, 119, 84–90, doi:10.1210/endo-119-1-84.
- Chertow, B.S.; Sivitz, W.I.; Baranetsky, N.G.; Clark, S.A.; Waite, A.; Deluca, H.F. Cellular mechanisms of insulin release: The effects of vitamin D deficiency and repletion on rat insulin secretion. Endocrinology 1983, 113, 1511–1518, doi:10.1210/endo-113-4-1511.
- Norman, A.W.; Frankel, J.B.; Heldt, A.M.; Grodsky, G.M. Vitamin D deficiency inhibits pancreatic secretion of insulin. Science 1980, 209, 823–825.
- Tanaka, Y.; Seino, Y.; Ishida, M.; Yamaoka, K.; Yabuuchi, H.; Ishida, H.; Seino, S.; Seino, Y.; Imura, H. Effect of vitamin D3 on the pancreatic secretion of insulin and somatostatin. Acta Endocrinol. 1984, 105, 528–533.
- Kadowaki, S.; Norman, A.W. Dietary vitamin D is essential for normal insulin secretion from the perfused rat pancreas. J. Clin. Investig. 1984, 73, 759–766, doi:10.1172/JCI111269.
- Mitri, J.; Pittas, A.G. Vitamin D and diabetes. Endocrinol. Metab. Clin. N. Am. 2014, 43, 205–232, doi:10.1016/j.ecl.2013.09.010.
- Inomata, S.; Kadowaki, S.; Yamatani, T.; Fukase, M.; Fujita, T. Effect of 1 alpha (OH)-vitamin D3 on insulin secretion in diabetes mellitus. Bone Miner. 1986, 1, 187–192.
- Boucher, B.J.; Mannan, N.; Noonan, K.; Hales, C.N.; Evans, S.J. Glucose intolerance and impairment of insulin secretion in relation to vitamin D deficiency in east London Asians. Diabetologia 1995, 38, 1239–1245.
- Johnson, J.A.; Grande, J.P.; Roche, P.C.; Kumar, R. Immunohistochemical localization of the 1,25(OH)2D3 receptor and calbindin D28k in human and rat pancreas. Am. J. Physiol. 1994, 267, E356–E360, doi:10.1152/ajpendo.1994.267.3.E356.
- Borissova, A.M.; Tankova, T.; Kirilov, G.; Dakovska, L.; Kovacheva, R. The effect of vitamin D3 on insulin secretion and peripheral insulin sensitivity in type 2 diabetic patients. Int. J. Clin. Pract. 2003, 57, 258–261.
- Nyomba, B.L.; Auwerx, J.; Bormans, V.; Peeters, T.L.; Pelemans, W.; Reynaert, J.; Bouillon, R.; Vantrappen, G.; De Moor, P. Pancreatic secretion in man with subclinical vitamin D deficiency. Diabetologia 1986, 29, 34–38.
- Altieri, B.; Grant, W.B.; Della Casa, S.; Orio, F.; Pontecorvi, A.; Colao, A.; Sarno, G.; Muscogiuri, G. Vitamin D and pancreas: The role of sunshine vitamin in the pathogenesis of diabetes mellitus and pancreatic cancer. Crit. Rev. Food Sci. Nutr. 2017, 57, 3472–3488, doi:10.1080/10408398.2015.1136922.
- Doyle, M.E.; Egan, J.M. Pharmacological agents that directly modulate insulin secretion. Pharmacol. Rev. 2003, 55, 105–131, doi:10.1124/pr.55.1.7.
- Santos, G.J. dos; Ferreira, S.M.; Ortis, F.; Rezende, L.F.; Li, C.; Naji, A.; Carneiro, E.M.; Kaestner, K.H.; Boschero, A.C. Metabolic memory of ß-cells controls insulin secretion and is mediated by CaMKII. Mol. Metab. 2014, 3, 484–489, doi:10.1016/j.molmet.2014.03.011.
- Dalle, S.; Quoyer, J.; Varin, E.; Costes, S. Roles and regulation of the transcription factor CREB in pancreatic β-cells. Curr. Mol. Pharmacol. 2011, 4, 187–195.
- Sooy, K.; Schermerhorn, T.; Noda, M.; Surana, M.; Rhoten, W.B.; Meyer, M.; Fleischer, N.; Sharp, G.W.; Christakos, S. Calbindin-D(28k) controls [Ca(2+)](i) and insulin release. Evidence obtained from calbindin-d(28k) knockout mice and beta cell lines. J. Biol. Chem. 1999, 274, 34343–34349.
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.-C.; Jurutka, P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2013, 92, 77–98, doi:10.1007/s00223-012-9619-0.
- Wasserman, R.H. Vitamin D and the dual processes of intestinal calcium absorption. J. Nutr. 2004, 134, 3137–3139, doi:10.1093/jn/134.11.3137.
- Viragh, P.A. de; Haglid, K.G.; Celio, M.R. Parvalbumin increases in the caudate putamen of rats with vitamin D hypervitaminosis. Proc. Natl. Acad. Sci. USA 1989, 86, 3887–3890, doi:10.1073/pnas.86.10.3887.
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776, doi:10.1210/er.2008-0004.
- Ni, Z.; Smogorzewski, M.; Massry, S.G. Effects of parathyroid hormone on cytosolic calcium of rat adipocytes. Endocrinology 1994, 135, 1837–1844, doi:10.1210/endo.135.5.7525254.
- Baczynski, R.; Massry, S.G.; Magott, M.; el-Belbessi, S.; Kohan, R.; Brautbar, N. Effect of parathyroid hormone on energy metabolism of skeletal muscle. Kidney Int. 1985, 28, 722–727.
- Reusch, J.E.; Begum, N.; Sussman, K.E.; Draznin, B. Regulation of GLUT-4 phosphorylation by intracellular calcium in adipocytes. Endocrinology 1991, 129, 3269–3273, doi:10.1210/endo-129-6-3269.
- Thomas, D.M.; Rogers, S.D.; Sleeman, M.W.; Pasquini, G.M.; Bringhurst, F.R.; Ng, K.W.; Zajac, J.D.; Best, J.D. Modulation of glucose transport by parathyroid hormone and insulin in UMR 106-01, a clonal rat osteogenic sarcoma cell line. J. Mol. Endocrinol. 1995, 14, 263–275.
- Teegarden, D.; Donkin, S.S. Vitamin D: Emerging new roles in insulin sensitivity. Nutr. Res. Rev. 2009, 22, 82–92, doi:10.1017/S0954422409389301.
- Sung, C.-C.; Liao, M.-T.; Lu, K.-C.; Wu, C.-C. Role of vitamin D in insulin resistance. J. Biomed. Biotechnol. 2012, 2012, 634195, doi:10.1155/2012/634195.
- Chiu, K.C.; Chuang, L.M.; Lee, N.P.; Ryu, J.M.; McGullam, J.L.; Tsai, G.P.; Saad, M.F. Insulin sensitivity is inversely correlated with plasma intact parathyroid hormone level. Metab. Clin. Exp. 2000, 49, 1501–1505, doi:10.1053/meta.2000.17708.
- Reis, J.P.; von Mühlen, D.; Kritz-Silverstein, D.; Wingard, D.L.; Barrett-Connor, E. Vitamin D, parathyroid hormone levels, and the prevalence of metabolic syndrome in community-dwelling older adults. Diabetes Care 2007, 30, 1549–1555, doi:10.2337/dc06-2438.
- Wright, D.C.; Hucker, K.A.; Holloszy, J.O.; Han, D.H. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes 2004, 53, 330–335, doi:10.2337/diabetes.53.2.330.
- Muscogiuri, G.; Chavez, A.O.; Gastaldelli, A.; Perego, L.; Tripathy, D.; Saad, M.J.; Velloso, L.; Folli, F. The crosstalk between insulin and renin-angiotensin-aldosterone signaling systems and its effect on glucose metabolism and diabetes prevention. Curr. Vasc. Pharmacol. 2008, 6, 301–312.
- Wei, Y.; Sowers, J.R.; Clark, S.E.; Li, W.; Ferrario, C.M.; Stump, C.S. Angiotensin II-induced skeletal muscle insulin resistance mediated by NF-kappaB activation via NADPH oxidase. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E345–E351, doi:10.1152/ajpendo.00456.2007.
- Yuan, W.; Pan, W.; Kong, J.; Zheng, W.; Szeto, F.L.; Wong, K.E.; Cohen, R.; Klopot, A.; Zhang, Z.; Li, Y.C. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J. Biol. Chem. 2007, 282, 29821–29830, doi:10.1074/jbc.M705495200.
- Kong, J.; Li, Y.C. Effect of ANG II type I receptor antagonist and ACE inhibitor on vitamin D receptor-null mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R255–R261, doi:10.1152/ajpregu.00517.2002.
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.-F.; Liu, S.Q.; Cao, L.-P. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238, doi:10.1172/JCI15219.
- Angellotti, E.; Pittas, A.G. The Role of Vitamin D in the Prevention of Type 2 Diabetes: To D or Not to D? Endocrinology 2017, 158, 2013–2021, doi:10.1210/en.2017-00265.
- Simpson, R.U.; Thomas, G.A.; Arnold, A.J. Identification of 1,25-dihydroxyvitamin D3 receptors and activities in muscle. J. Biol. Chem. 1985, 260, 8882–8891.
- Maestro, B.; Campión, J.; Dávila, N.; Calle, C. Stimulation by 1,25-dihydroxyvitamin D3 of insulin receptor expression and insulin responsiveness for glucose transport in U-937 human promonocytic cells. Endocr. J. 2000, 47, 383–391.
- Dunlop, T.W.; Väisänen, S.; Frank, C.; Molnár, F.; Sinkkonen, L.; Carlberg, C. The human peroxisome proliferator-activated receptor delta gene is a primary target of 1alpha,25-dihydroxyvitamin D3 and its nuclear receptor. J. Mol. Biol. 2005, 349, 248–260, doi:10.1016/j.jmb.2005.03.060.
- Manna, P.; Achari, A.E.; Jain, S.K. 1,25(OH)2-vitamin D3 upregulates glucose uptake mediated by SIRT1/IRS1/GLUT4 signaling cascade in C2C12 myotubes. Mol. Cell. Biochem. 2018, 444, 103–108, doi:10.1007/s11010-017-3235-2.
- Bland, R.; Markovic, D.; Hills, C.E.; Hughes, S.V.; Chan, S.L.F.; Squires, P.E.; Hewison, M. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in pancreatic islets. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 121–125, doi:10.1016/j.jsbmb.2004.03.115.
- Zeitz, U.; Weber, K.; Soegiarto, D.W.; Wolf, E.; Balling, R.; Erben, R.G. Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. FASEB J. 2003, 17, 509–511, doi:10.1096/fj.02-0424fje.
- Maestro, B.; Molero, S.; Bajo, S.; Dávila, N.; Calle, C. Transcriptional activation of the human insulin receptor gene by 1,25-dihydroxyvitamin D(3). Cell Biochem. Funct. 2002, 20, 227–232, doi:10.1002/cbf.951.
- Green, T.J.; Skeaff, C.M.; Rockell, J.E.P.; Venn, B.J.; Lambert, A.; Todd, J.; Khor, G.L.; Loh, S.P.; Muslimatun, S.; Agustina, R.; et al. Vitamin D status and its association with parathyroid hormone concentrations in women of child-bearing age living in Jakarta and Kuala Lumpur. Eur. J. Clin. Nutr. 2008, 62, 373–378, doi:10.1038/sj.ejcn.1602696.
- Maestro, B.; Dávila, N.; Carranza, M.C.; Calle, C. Identification of a Vitamin D response element in the human insulin receptor gene promoter. J. Steroid Biochem. Mol. Biol. 2003, 84, 223–230.
- George, N.; Kumar, T.P.; Antony, S.; Jayanarayanan, S.; Paulose, C.S. Effect of vitamin D3 in reducing metabolic and oxidative stress in the liver of streptozotocin-induced diabetic rats. Br. J. Nutr. 2012, 108, 1410–1418, doi:10.1017/S0007114511006830.
- Alkharfy, K.M.; Al-Daghri, N.M.; Yakout, S.M.; Hussain, T.; Mohammed, A.K.; Krishnaswamy, S. Influence of vitamin D treatment on transcriptional regulation of insulin-sensitive genes. Metab. Syndr. Relat. Disord. 2013, 11, 283–288, doi:10.1089/met.2012.0068.
- Calle, C.; Maestro, B.; García-Arencibia, M. Genomic actions of 1,25-dihydroxyvitamin D3 on insulin receptor gene expression, insulin receptor number and insulin activity in the kidney, liver and adipose tissue of streptozotocin-induced diabetic rats. BMC Mol. Biol. 2008, 9, 65, doi:10.1186/1471-2199-9-65.
- Chang, Y.-C.; Chuang, L.-M. The role of oxidative stress in the pathogenesis of type 2 diabetes: From molecular mechanism to clinical implication. Am. J. Transl. Res. 2010, 2, 316–331.
- Garbossa, S.G.; Folli, F. Vitamin D, sub-inflammation and insulin resistance. A window on a potential role for the interaction between bone and glucose metabolism. Rev. Endocr. Metab. Disord. 2017, 18, 243–258, doi:10.1007/s11154-017-9423-2.
- Ježek, P.; Dlasková, A.; Plecitá-Hlavatá, L. Redox homeostasis in pancreatic β cells. Oxid. Med. Cell. Longev. 2012, 2012, 932838, doi:10.1155/2012/932838.
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262, doi:10.1016/j.bj.2017.06.007.
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol., Cell. Physiol. 2019, 317, C420–C433, doi:10.1152/ajpcell.00141.2019.
- Solinas, G.; Karin, M. JNK1 and IKKbeta: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611, doi:10.1096/fj.09-151340.
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative Stress and the Etiology of Insulin Resistance and Type 2 Diabetes. Free Radic. Biol. Med. 2011, 51, 993–999, doi:10.1016/j.freeradbiomed.2010.12.005.
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945.
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I.H. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550, doi:10.1042/BCJ20160503C.
- Fridlyand, L.E.; Philipson, L.H. Reactive species and early manifestation of insulin resistance in type 2 diabetes. Diabetes Obes. Metab. 2006, 8, 136–145, doi:10.1111/j.1463-1326.2005.00496.x.
- Paolisso, G.; D’Amore, A.; Volpe, C.; Balbi, V.; Saccomanno, F.; Galzerano, D.; Giugliano, D.; Varricchio, M.; D’Onofrio, F. Evidence for a relationship between oxidative stress and insulin action in non-insulin-dependent (type II) diabetic patients. Metab. Clin. Exp. 1994, 43, 1426–1429.
- Nourooz-Zadeh, J.; Rahimi, A.; Tajaddini-Sarmadi, J.; Tritschler, H.; Rosen, P.; Halliwell, B.; Betteridge, D.J. Relationships between plasma measures of oxidative stress and metabolic control in NIDDM. Diabetologia 1997, 40, 647–653, doi:10.1007/s001250050729.
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are Oxidative Stress−Activated Signaling Pathways Mediators of Insulin Resistance and β-Cell Dysfunction? Diabetes 2003, 52, 1–8, doi:10.2337/diabetes.52.1.1.
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625, doi:10.2337/diabetes.54.6.1615.
- Rao, M.S.; Reddy, J.K. Peroxisomal β-Oxidation and Steatohepatitis. Semin. Liver Dis. 2001, 21, 043–056, doi:10.1055/s-2001-12928.
- Lowell, B.B.; Shulman, G.I. Mitochondrial Dysfunction and Type 2 Diabetes. Science 2005, 307, 384–387, doi:10.1126/science.1104343.
- Carlsson, C.; Håkan Borg, L.A.; Welsh, N. Sodium Palmitate Induces Partial Mitochondrial Uncoupling and Reactive Oxygen Species in Rat Pancreatic Islets in Vitro. Endocrinology 1999, 140, 3422–3428, doi:10.1210/endo.140.8.6908.
- Scaini, G.; Rezin, G.T.; Carvalho, A.F.; Streck, E.L.; Berk, M.; Quevedo, J. Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev. 2016, 68, 694–713, doi:10.1016/j.neubiorev.2016.06.040.
- Kim, H.K.; Andreazza, A.C.; Yeung, P.Y.; Isaacs-Trepanier, C.; Young, L.T. Oxidation and nitration in dopaminergic areas of the prefrontal cortex from patients with bipolar disorder and schizophrenia. J. Psychiatry Neurosci. 2014, 39, 276–285.
- Silvagno, F.; Consiglio, M.; Foglizzo, V.; Destefanis, M.; Pescarmona, G. Mitochondrial translocation of vitamin D receptor is mediated by the permeability transition pore in human keratinocyte cell line. PLoS ONE 2013, 8, e54716, doi:10.1371/journal.pone.0054716.
- Silvagno, F.; De Vivo, E.; Attanasio, A.; Gallo, V.; Mazzucco, G.; Pescarmona, G. Mitochondrial localization of vitamin D receptor in human platelets and differentiated megakaryocytes. PLoS ONE 2010, 5, e8670, doi:10.1371/journal.pone.0008670.
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediat. Inflamm. 2015, 2015, 105828, doi:10.1155/2015/105828.
- Qatanani, M.; Lazar, M.A. Mechanisms of obesity-associated insulin resistance: Many choices on the menu. Genes Dev. 2007, 21, 1443–1455, doi:10.1101/gad.1550907.
- Rains, J.L.; Jain, S.K. Oxidative Stress, Insulin Signaling and Diabetes. Free Radic. Biol. Med. 2011, 50, 567–575, doi:10.1016/j.freeradbiomed.2010.12.006.
- Ricciardi, C.J.; Bae, J.; Esposito, D.; Komarnytsky, S.; Hu, P.; Chen, J.; Zhao, L. 1,25-Dihydroxyvitamin D3/vitamin D receptor suppresses brown adipocyte differentiation and mitochondrial respiration. Eur. J. Nutr. 2015, 54, 1001–1012, doi:10.1007/s00394-014-0778-9.
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672, doi:10.3390/ijms19061672.
- Consiglio, M.; Viano, M.; Casarin, S.; Castagnoli, C.; Pescarmona, G.; Silvagno, F. Mitochondrial and lipogenic effects of vitamin D on differentiating and proliferating human keratinocytes. Exp. Dermatol. 2015, 24, 748–753, doi:10.1111/exd.12761.
- Sun, X.; Zemel, M.B. 1α,25-Dihydroxyvitamin D3 Modulation of Adipocyte Reactive Oxygen Species Production. Obesity 2007, 15, 1944–1953, doi:10.1038/oby.2007.232.
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71, doi:10.1016/j.bbrc.2015.01.008.
- Dong, J.; Wong, S.L.; Lau, C.W.; Lee, H.K.; Ng, C.F.; Zhang, L.; Yao, X.; Chen, Z.Y.; Vanhoutte, P.M.; Huang, Y. Calcitriol protects renovascular function in hypertension by down-regulating angiotensin II type 1 receptors and reducing oxidative stress. Eur. Heart J. 2012, 33, 2980–2990, doi:10.1093/eurheartj/ehr459.
- Briones, T.L.; Darwish, H. Retraction notice to “DECREASE IN AGE-RELATED TAU HYPERPHOSPHORYLATION AND COGNITIVE IMPROVEMENT FOLLOWING VITAMIN D SUPPLEMENTATION ARE ASSOCIATED WITH MODULATION OF BRAIN ENERGY METABOLISM AND REDOX STATE”. Neuroscience 2014, 262, 143–155, doi:10.1016/j.neuroscience.2013.12.064.
- Garcion, E.; Sindji, L.; Leblondel, G.; Brachet, P.; Darcy, F. 1,25-Dihydroxyvitamin D3 Regulates the Synthesis of γ-Glutamyl Transpeptidase and Glutathione Levels in Rat Primary Astrocytes. J. Neurochem. 1999, 73, 859–866, doi:10.1046/j.1471-4159.1999.0730859.x.
- Bao, B.-Y.; Ting, H.-J.; Hsu, J.-W.; Lee, Y.-F. Protective role of 1α, 25-dihydroxyvitamin D3 against oxidative stress in nonmalignant human prostate epithelial cells. Int. J. Cancer 2008, 122, 2699–2706, doi:10.1002/ijc.23460.
- Jain, S.K.; Micinski, D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem. Biophys. Res. Commun. 2013, 437, 7–11, doi:10.1016/j.bbrc.2013.06.004.