The redox-active TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl-4-yl) fragment is a popular component of organic energy storage and catalytic systems as its benefits include remarkable electrochemical performance and decent physical properties. TEMPO is a verstile compound that finds its use in various chemical and biological systems, and is also known to be an efficient catalyst for alcohol oxidation, oxygen reduction, and various complex organic reactions. It can be attached to various aliphatic and conductive polymers to form energy storage compounds for organic batteries or high-loading catalysis systems. The performance and efficiency of TEMPO-containing materials strongly depend on the molecular structure, and thus rational design of such compounds is vital for successful implementation.
- TEMPO
- nitroxyl
- stable radicals
- redox polymers
- conductive polymers
- organic radical polymers
- charge transfer mechanisms
1. Introduction
2. Electroactive Materials Based on TEMPO
2.1. Synthetic Approaches
2.1.1. Non-Conductive Backbones
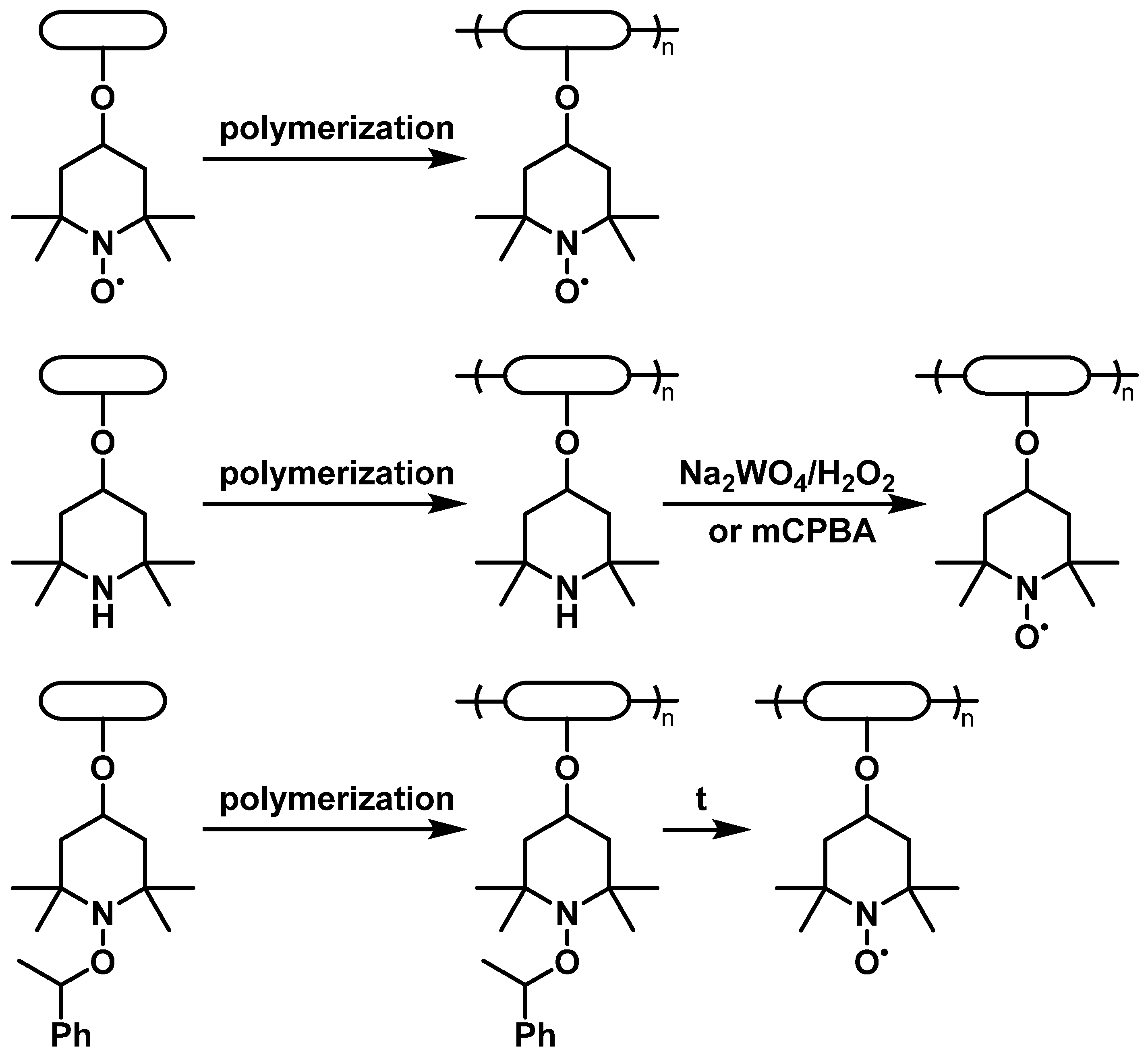
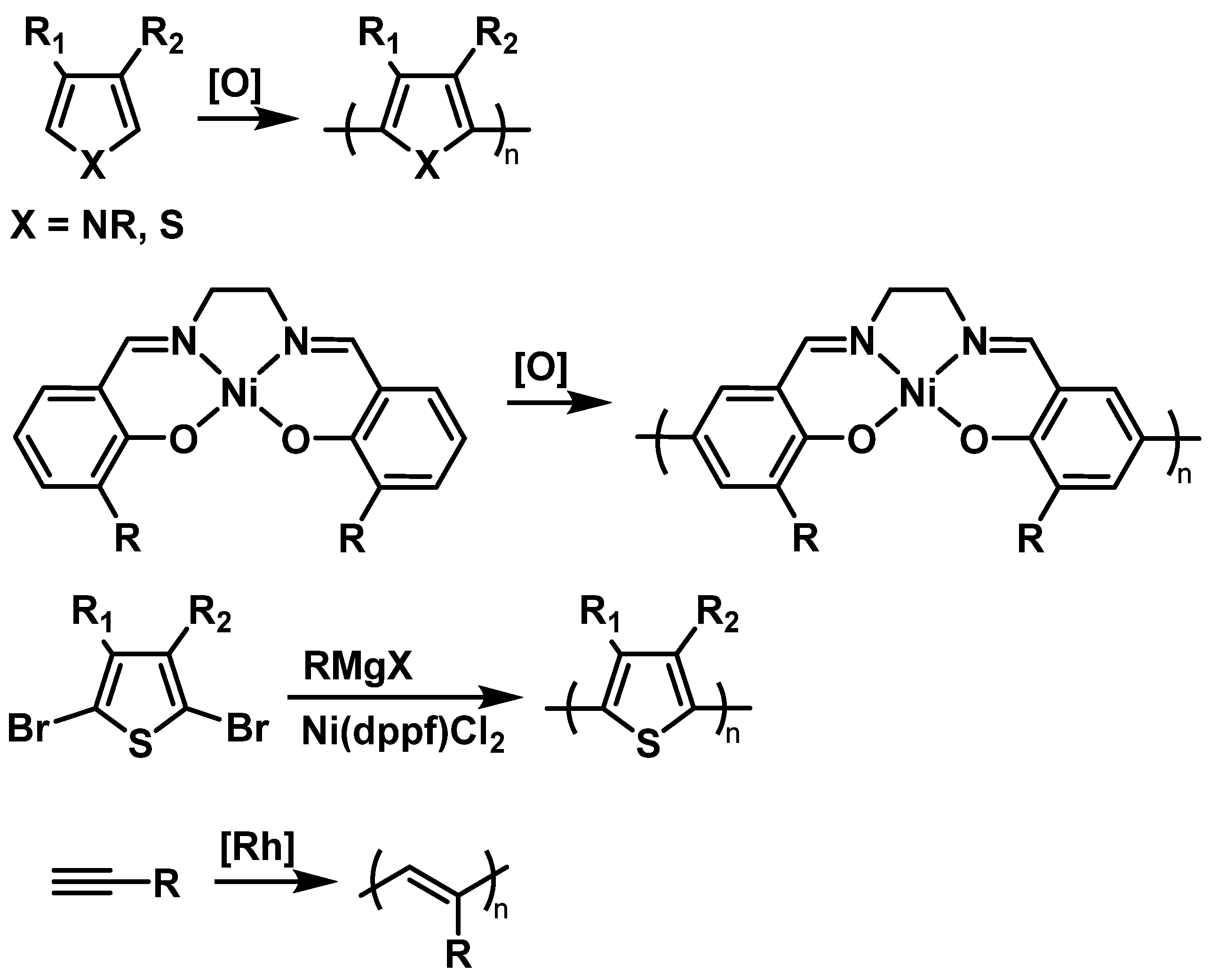
2.1.2. Conductive Backbones
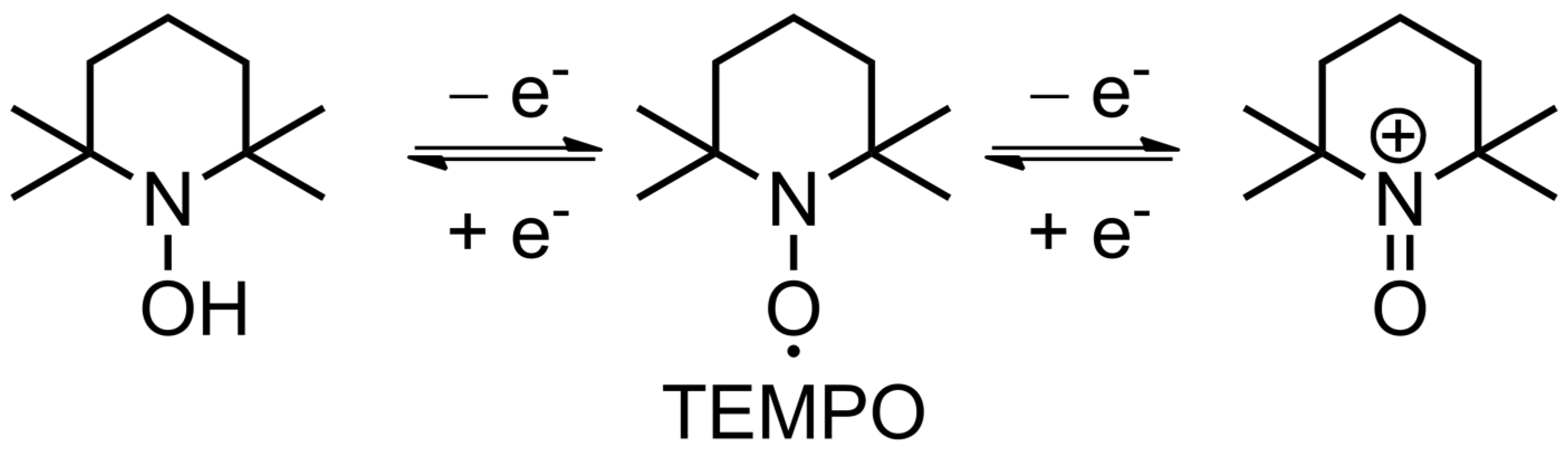
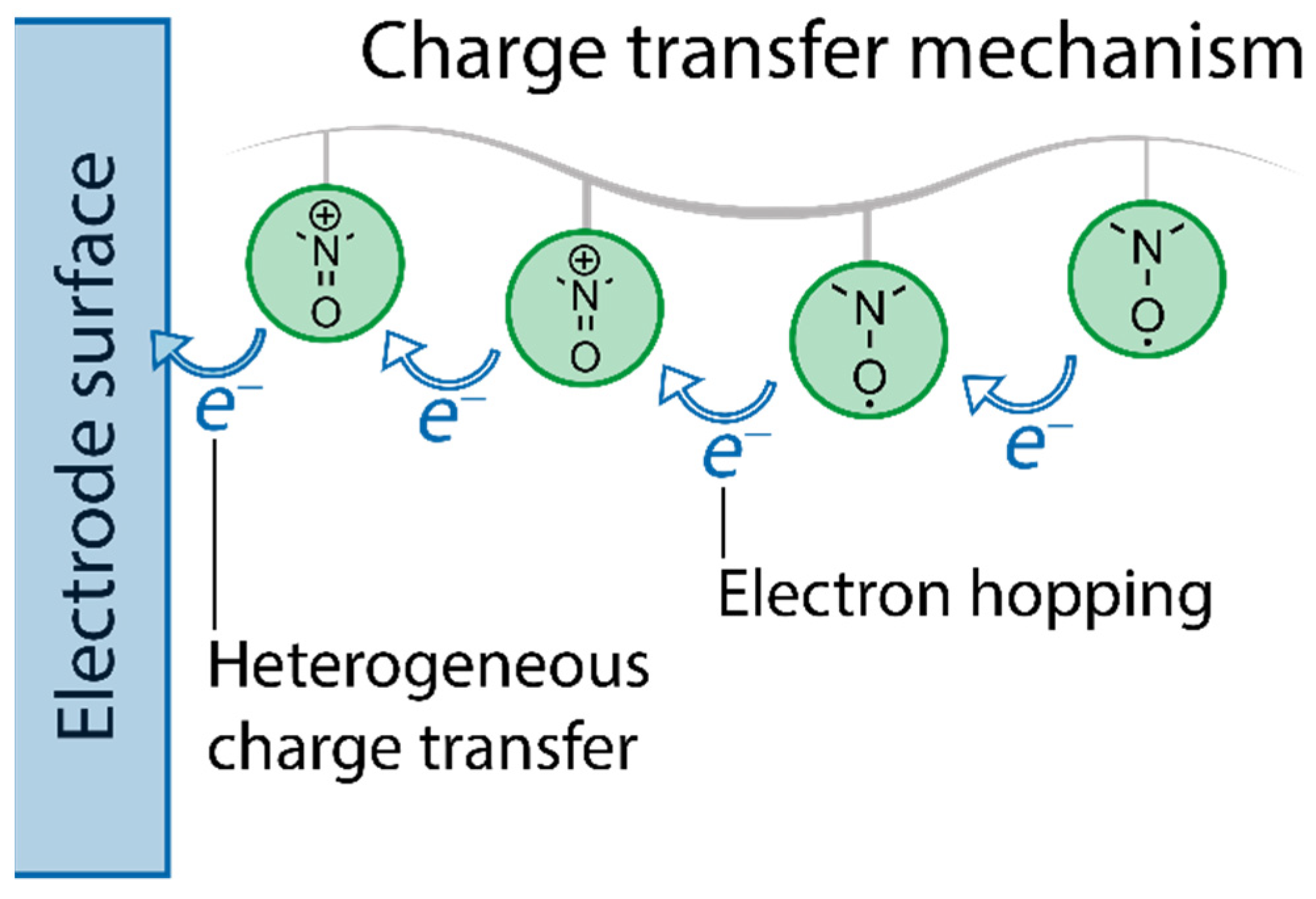
2.2. Fundamental Electrochemistry of TEMPO Transformations
This entry is adapted from the peer-reviewed paper 10.3390/en15072699
References
- Fan, E.; Li, L.; Wang, Z.; Lin, J.; Huang, Y.; Yao, Y.; Chen, R.; Wu, F. Sustainable Recycling Technology for Li-Ion Batteries and Beyond: Challenges and Future Prospects. Chem. Rev. 2020, 120, 7020–7063.
- Banza Lubaba Nkulu, C.; Casas, L.; Haufroid, V.; De Putter, T.; Saenen, N.D.; Kayembe-Kitenge, T.; Musa Obadia, P.; Kyanika Wa Mukoma, D.; Lunda Ilunga, J.M.; Nawrot, T.S.; et al. Sustainability of artisanal mining of cobalt in DR Congo. Nat. Sustain. 2018, 1, 495–504.
- Poizot, P.; Gaubicher, J.; Renault, S.; Dubois, L.; Liang, Y.; Yao, Y. Opportunities and Challenges for Organic Electrodes in Electrochemical Energy Storage. Chem. Rev. 2020, 120, 6490–6557.
- Xie, Y.; Zhang, K.; Yamauchi, Y.; Oyaizu, K.; Jia, Z. Nitroxide radical polymers for emerging plastic energy storage and organic electronics: Fundamentals, materials, and applications. Mater. Horiz. 2021, 8, 803–829.
- Esser, B.; Dolhem, F.; Becuwe, M.; Poizot, P.; Vlad, A.; Brandell, D. A perspective on organic electrode materials and technologies for next generation batteries. J. Power Sources 2021, 482, 228814.
- Goujon, N.; Casado, N.; Patil, N.; Marcilla, R.; Mecerreyes, D. Organic batteries based on just redox polymers. Prog. Polym. Sci. 2021, 122, 101449.
- Rohland, P.; Schröter, E.; Nolte, O.; Newkome, G.R.; Hager, M.D.; Schubert, U.S. Redox-active polymers: The magic key towards energy storage—A polymer design guideline progress in polymer science. Prog. Polym. Sci. 2022, 125, 101474.
- Tan, Y.; Hsu, S.N.; Tahir, H.; Dou, L.; Savoie, B.M.; Boudouris, B.W. Electronic and Spintronic Open-Shell Macromolecules, Quo Vadis? J. Am. Chem. Soc. 2022, 144, 626–647.
- Hansen, K.A.; Blinco, J.P. Nitroxide radical polymers—A versatile material class for high-tech applications. Polym. Chem. 2018, 9, 1479–1516.
- Muench, S.; Wild, A.; Friebe, C.; Haupler, B.; Janoschka, T.; Schubert, U.S. Polymer-Based Organic Batteries. Chem. Rev. 2016, 116, 9438–9484.
- Janoschka, T.; Hager, M.D.; Schubert, U.S. Powering up the Future: Radical Polymers for Battery Applications. Adv. Mater. 2012, 24, 6397–6409.
- Chen, Y.; Zhuo, S.; Li, Z.; Wang, C. Redox polymers for rechargeable metal-ion batteries. EnergyChem 2020, 2, 100030.
- Kim, K.; Baldaguez Medina, P.; Elbert, J.; Kayiwa, E.; Cusick, R.D.; Men, Y.; Su, X. Molecular Tuning of Redox-Copolymers for Selective Electrochemical Remediation. Adv. Funct. Mater. 2020, 30, 2004635.
- Mohebbati, N.; Prudlik, A.; Scherkus, A.; Gudkova, A.; Francke, R. TEMPO-Modified Polymethacrylates as Mediators in Electrosynthesis—Redox Behavior and Electrocatalytic Activity toward Alcohol Substrates. ChemElectroChem 2021, 8, 3837–3843.
- Yang, S.G.; Park, H.J.; Kim, J.W.; Jung, J.M.; Kim, M.J.; Jegal, H.G.; Kim, I.S.; Kang, M.J.; Wee, G.; Yang, H.Y.; et al. Mito-TEMPO improves development competence by reducing superoxide in preimplantation porcine embryos. Sci. Rep. 2018, 8, 10130.
- Shetty, S.; Kumar, R.; Bharati, S. Mito-TEMPO, a mitochondria-targeted antioxidant, prevents N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Free Radic. Biol. Med. 2019, 136, 76–86.
- Komaba, S.; Tanaka, T.; Ozeki, T.; Taki, T.; Watanabe, H.; Tachikawa, H. Fast redox of composite electrode of nitroxide radical polymer and carbon with polyacrylate binder. J. Power Sources 2010, 195, 6212–6217.
- Bugnon, L.; Morton, C.J.H.; Novak, P.; Vetter, J.; Nesvadba, P. Synthesis of Poly(4-methacryloyloxy-TEMPO) via Group-Transfer Polymerization and Its Evaluation in Organic Radical Battery. Chem. Mater. 2007, 19, 2910–2914.
- Koshika, K.; Chikushi, N.; Sano, N.; Oyaizu, K.; Nishide, H. A TEMPO-substituted polyacrylamide as a new cathode material: An organic rechargeable device composed of polymer electrodes and aqueous electrolyte. Green Chem. 2010, 12, 1573–1575.
- Suguro, M.; Iwasa, S.; Kusachi, Y.; Morioka, Y.; Nakahara, K. Cationic Polymerization of Poly(vinyl ether) Bearing a TEMPO Radical: A New Cathode-Active Material for Organic Radical Batteries. Macromol. Rapid Commun. 2007, 28, 1929–1933.
- Sertkol, S.B.; Sinirlioglu, D.; Esat, B.; Muftuoglu, A.E. A novel cathode material based on polystyrene with pendant TEMPO moieties obtained via click reaction and its use in rechargeable batteries. J. Polym. Res. 2015, 22, 136.
- Nakahara, K.; Iriyama, J.; Iwasa, S.; Suguro, M.; Satoh, M.; Cairns, E.J. Cell properties for modified PTMA cathodes of organic radical batteries. J. Power Sources 2007, 165, 398–402.
- Aqil, M.; Ouhib, F.; Aqil, A.; El Idrissi, A.; Detrembleur, C.; Jérôme, C. Polymer ionic liquid bearing radicals as an active material for organic batteries with ultrafast charge-discharge rate. Eur. Polym. J. 2018, 106, 242–248.
- Suga, T.; Yoshimura, K.; Nishide, H. Nitroxide-Substituted Polyether as a New Material for Batteries. Macromol. Symp. 2006, 245–246, 416–422.
- Qu, J.; Katsumata, T.; Satoh, M.; Wada, J.; Masuda, T. Poly(7-oxanorbornenes) carrying 2,2,6,6-tetramethylpiperidine-1-oxy (TEMPO) radicals: Synthesis and charge/discharge properties. Polymer 2009, 50, 391–396.
- Ibe, T.; Frings, R.B.; Lachowicz, A.; Kyo, S.; Nishide, H. Nitroxide polymer networks formed by Michael addition: On site-cured electrode-active organic coating. Chem. Commun. 2010, 46, 3475–3477.
- Sukegawa, T.; Sato, K.; Oyaizu, K.; Nishide, H. Efficient charge transport of a radical polyether/SWCNT composite electrode for an organic radical battery with high charge-storage density. RSC Adv. 2015, 5, 15448–15452.
- Sukegawa, T.; Masuko, I.; Oyaizu, K.; Nishide, H. Expanding the Dimensionality of Polymers Populated with Organic Robust Radicals toward Flow Cell Application: Synthesis of TEMPO-Crowded Bottlebrush Polymers Using Anionic Polymerization and ROMP. Macromolecules 2014, 47, 8611–8617.
- Zhu, J.; Zhu, T.; Tuo, H.; Zhang, W. Synthesis of a TEMPO-Substituted Polyacrylamide Bearing a Sulfonate Sodium Pendant and Its Properties in an Organic Radical Battery. Polymers 2019, 11, 2076.
- Hickey, D.P.; Milton, R.D.; Chen, D.; Sigman, M.S.; Minteer, S.D. TEMPO-Modified Linear Poly(ethylenimine) for Immobilization-Enhanced Electrocatalytic Oxidation of Alcohols. ACS Catal. 2015, 5, 5519–5524.
- Chen, Y.; Zhang, Y.; Liu, X.; Fan, X.; Bai, B.; Yang, K.; Liang, Z.; Zhang, Z.; Mai, K. Long-Life and High-Power Binder-Free Cathode Based on One-Step Synthesis of Radical Polymers with Multi-Pendant Groups. Macromol. Rapid Commun. 2018, 39, 1800195.
- Qu, J.; Khan, F.Z.; Satoh, M.; Wada, J.; Hayashi, H.; Mizoguchi, K.; Masuda, T. Synthesis and charge/discharge properties of cellulose derivatives carrying free radicals. Polymer 2008, 49, 1490–1496.
- Aydın, M.; Esat, B.; Kılıç, Ç.; Köse, M.E.; Ata, A.; Yılmaz, F. A polythiophene derivative bearing TEMPO as a cathode material for rechargeable batteries. Eur. Polym. J. 2011, 47, 2283–2294.
- Xu, L.; Ji, L.; Wang, G.; Zhang, C.; Su, C. A novel nitroxide radical polymer-containing conductive polyaniline as molecular skeleton: Its synthesis and electrochemical properties as organic cathode. Ionics 2016, 22, 1377–1385.
- Zhang, Y.; Park, A.M.; McMillan, S.R.; Harmon, N.J.; Flatté, M.E.; Fuchs, G.D.; Ober, C.K. Charge Transport in Conjugated Polymers with Pendent Stable Radical Groups. Chem. Mater. 2018, 30, 4799–4807.
- Qu, J.; Fujii, T.; Katsumata, T.; Suzuki, Y.; Shiotsuki, M.; Sanda, F.; Satoh, M.; Wada, J.; Masuda, T. Helical polyacetylenes carrying 2,2,6,6-tetramethyl-1-piperidinyloxy and 2,2,5,5-tetramethyl-1-pyrrolidinyloxy moieties: Their synthesis, properties, and function. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 5431–5445.
- Xu, L.; Yang, F.; Su, C.; Ji, L.; Zhang, C. Synthesis and properties of novel TEMPO-contained polypyrrole derivatives as the cathode material of organic radical battery. Electrochim. Acta 2014, 130, 148–155.
- Yi, J.; Tang, D.; Song, D.; Wu, X.; Shen, Z.; Li, M. Selective oxidation of benzyl alcohol on poly(4-(3-(pyrrol-1-yl)propionamido)-2,2,6,6-tetramethylpiperidin-1-yloxy) electrode. J. Solid State Electrochem. 2015, 19, 2291–2297.
- Li, F.; Gore, D.N.; Wang, S.; Lutkenhaus, J.L. Unusual Internal Electron Transfer in Conjugated Radical Polymers. Angew. Chem. 2017, 56, 9856–9859.
- Schwartz, P.O.; Pejic, M.; Wachtler, M.; Bäuerle, P. Synthesis and characterization of electroactive PEDOT-TEMPO polymers as potential cathode materials in rechargeable batteries. Synth. Met. 2018, 243, 51–57.
- Su, C.; Yang, F.; Xu, L.; Zhu, X.; He, H.; Zhang, C. Radical Polymer Containing a Polytriphenylamine Backbone: Its Synthesis and Electrochemical Performance as the Cathode of Lithium-Ion Batteries. Chempluschem 2015, 80, 606–611.
- Vereshchagin, A.A.; Lukyanov, D.A.; Kulikov, I.R.; Panjwani, N.A.; Alekseeva, E.A.; Behrends, J.; Levin, O.V. The Fast and the Capacious: A -TEMPO Redox-Conducting Polymer for Organic Batteries. Batter. Supercaps 2021, 4, 336–346.
- Nakahara, K.; Iwasa, S.; Satoh, M.; Morioka, Y.; Iriyama, J.; Suguro, M.; Hasegawa, E. Rechargeable batteries with organic radical cathodes. Chem. Phys. Lett. 2002, 359, 351–354.
- Nevers, D.R.; Brushett, F.R.; Wheeler, D.R. Engineering radical polymer electrodes for electrochemical energy storage. J. Power Sources 2017, 352, 226–244.
- Takahashi, K.; Korolev, K.; Tsuji, K.; Oyaizu, K.; Nishide, H.; Bryuzgin, E.; Navrotskiy, A.; Novakov, I. Facile grafting-onto-preparation of block copolymers of TEMPO and glycidyl methacrylates on an oxide substrate as an electrode-active layer. Polymer 2015, 68, 310–314.
- Koshika, K.; Sano, N.; Oyaizu, K.; Nishide, H. An ultrafast chargeable polymer electrode based on the combination of nitroxide radical and aqueous electrolyte. Chem. Commun. 2009, 7, 836–838.
- Nishide, H.; Suga, T. Organic Radical Battery. Electrochem. Soc. Interface 2005, 14, 32–36.
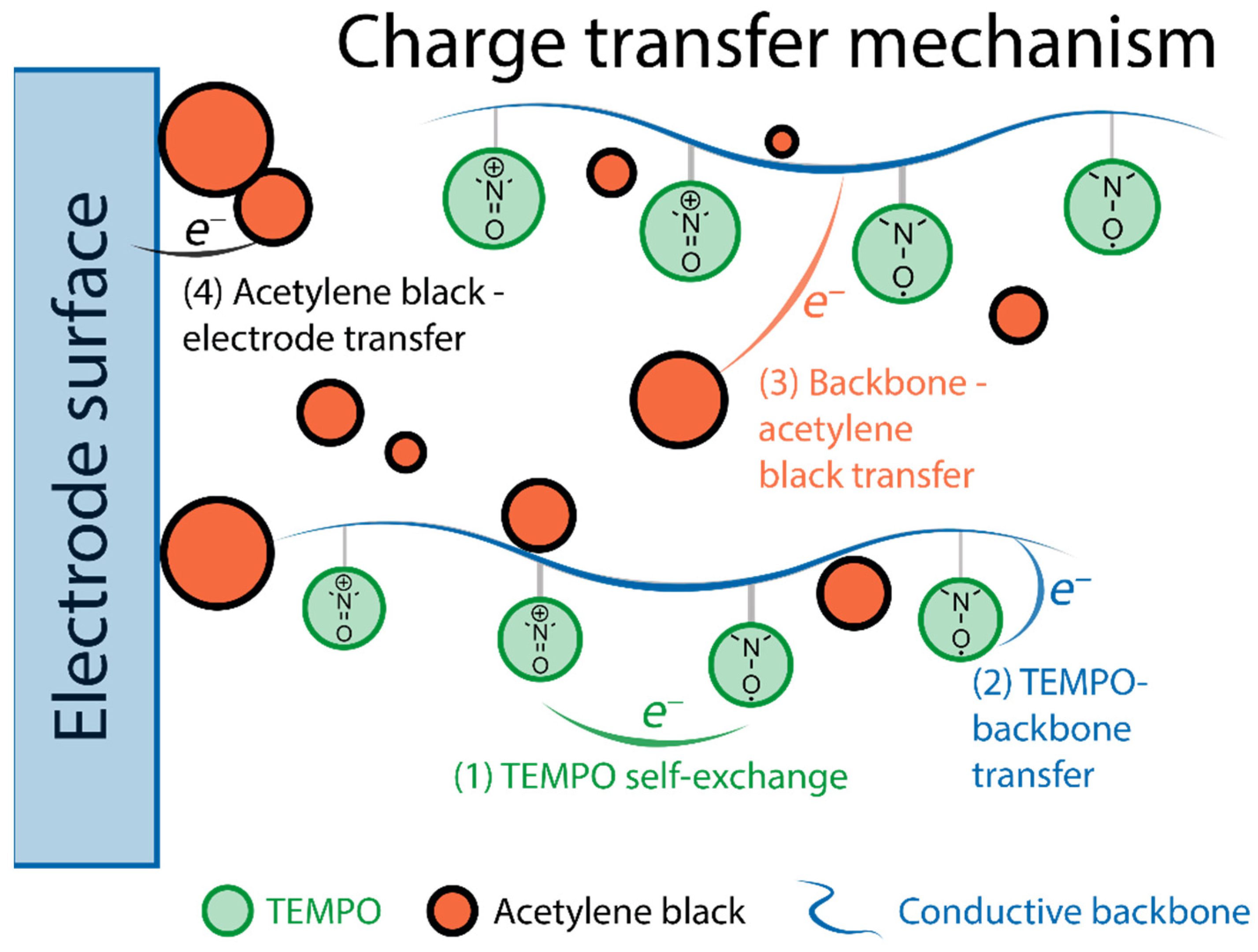
- Wang, S.; Easley, A.D.; Thakur, R.M.; Ma, T.; Yun, J.; Zhang, Y.; Ober, C.K.; Lutkenhaus, J.L. Quantifying internal charge transfer and mixed ion-electron transfer in conjugated radical polymers. Chem. Sci. 2020, 11, 9962–9970.
- Kemper, T.W.; Larsen, R.E.; Gennett, T. Relationship between Molecular Structure and Electron Transfer in a Polymeric Nitroxyl-Radical Energy Storage Material. J. Phys. Chem. C 2014, 118, 17213–17220.
- Oyaizu, K.; Ando, Y.; Konishi, H.; Nishide, H. Nernstian Adsorbate-like Bulk Layer of Organic Radical Polymers for High-Density Charge Storage Purposes. J. Am. Chem. Soc. 2008, 130, 14459–14461.
- Suga, T.; Pu, Y.J.; Oyaizu, K.; Nishide, H. Electron-Transfer Kinetics of Nitroxide Radicals as an Electrode-Active Material. Bull. Chem. Soc. Jpn. 2004, 77, 2203–2204.
- Nakahara, K.; Oyaizu, K.; Nishide, H. Organic Radical Battery Approaching Practical Use. Chem. Lett. 2011, 40, 222–227.
- Chatgilialoglu, C.; Studer, A. Encyclopedia of Radicals in Chemistry, Biology, and Materials; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2012; Volume 2.
- Grampp, G.; Rasmussen, K. Solvent dynamical effects on the electron self-exchange rate of the TEMPO/TEMPO+ couple (TEMPO = 2,2,6,6-tetramethyl-1-piperidinyloxy radical) Part I. ESR-linebroadening measurements at T = 298 K. Phys. Chem. Chem. Phys. 2002, 4, 5546–5549.
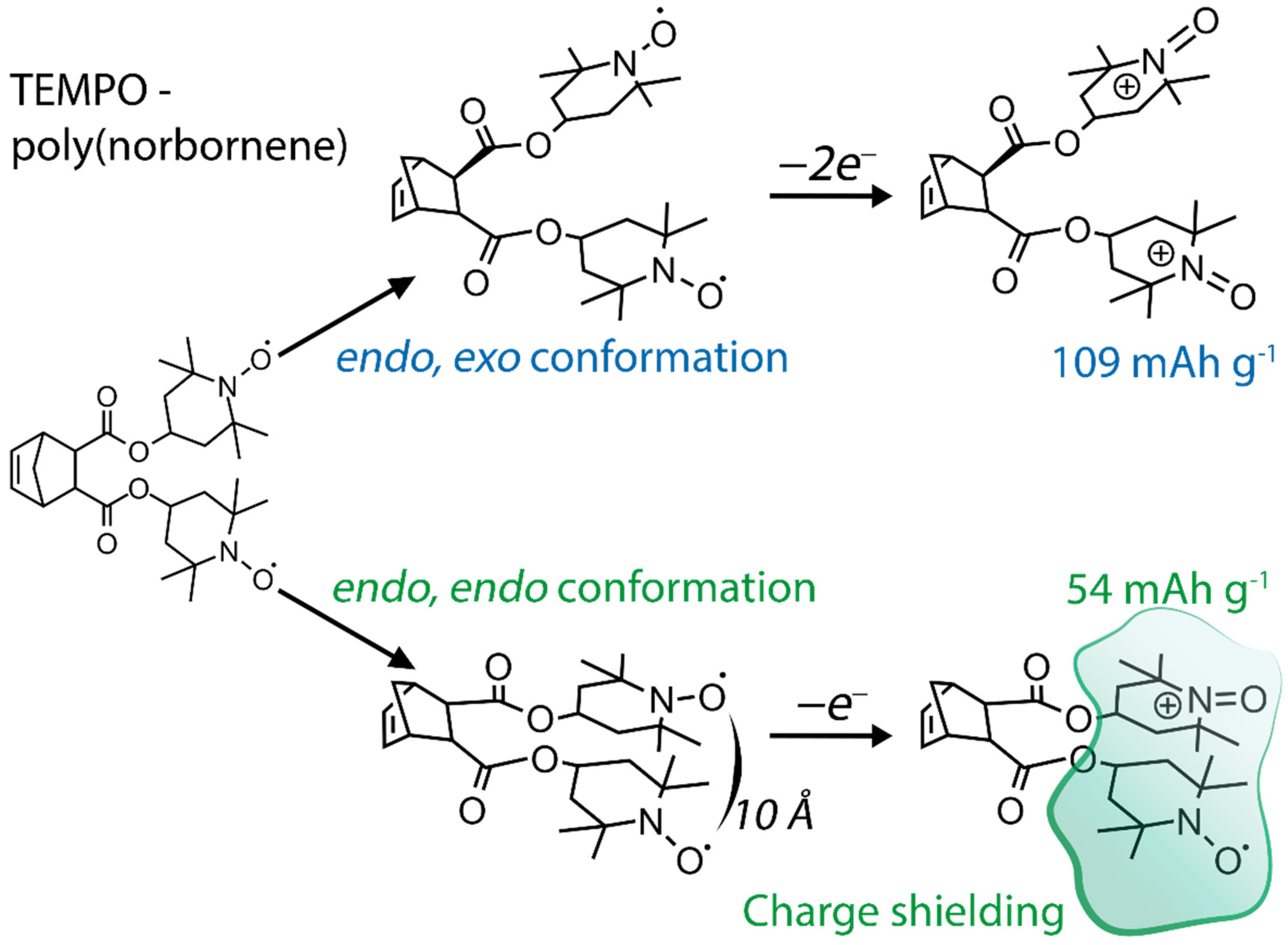
- Martin, H.J.; Hughes, B.K.; Braunecker, W.A.; Gennett, T.; Dadmun, M.D. The impact of radical loading and oxidation on the conformation of organic radical polymers by small angle neutron scattering. J. Mater. Chem. A 2018, 6, 15659–15667.
- Katsumata, T.; Satoh, M.; Wada, J.; Shiotsuki, M.; Sanda, F.; Masuda, T. Polyacetylene and Polynorbornene Derivatives Carrying TEMPO. Synthesis and Properties as Organic Radical Battery Materials. Macromol. Rapid Commun. 2006, 27, 1206–1211.
- Lutkenhaus, J. A radical advance for conducting polymers. Science 2018, 359, 1334–1335.
- Joo, Y.; Agarkar, V.; Sung, S.H.; Savoie, B.M.; Boudouris, B.W. A nonconjugated radical polymer glass with high electrical conductivity. Science 2018, 359, 1391–1395.
- Oka, K.; Nishide, H. Chapter 4 Radical Polymers for Rechargeable Batteries. In Redox Polymers for Energy and Nanomedicine; The Royal Society of Chemistry: London, UK, 2021; pp. 137–165.
- Tokue, H.; Oyaizu, K.; Sukegawa, T.; Nishide, H. TEMPO/Viologen Electrochemical Heterojunction for Diffusion-Controlled Redox Mediation: A Highly Rectifying Bilayer-Sandwiched Device Based on Cross-Reaction at the Interface between Dissimilar Redox Polymers. ACS Appl. Mater. Interfaces 2014, 6, 4043–4049.
- Suga, T.; Konishi, H.; Nishide, H. Photocrosslinked nitroxide polymer cathode-active materials for application in an organic-based paper battery. Chem. Commun. 2007, 17, 1730–1732.
- Ou, Y.; Zhang, Y.; Xiong, Y.; Hu, Z.; Dong, L. Three-dimensional porous radical polymer/reduced graphene oxide composite with two-electron redox reactions as high-performance cathode for lithium-ion batteries. Eur. Polym. J. 2021, 143, 110191.
- Stolze, C.; Janoschka, T.; Flauder, S.; Müller, F.A.; Hager, M.D.; Schubert, U.S. Investigation of Ice-Templated Porous Electrodes for Application in Organic Batteries. ACS Appl. Mater. Interfaces 2016, 8, 23614–23623.
- Xie, J.; Gu, P.; Zhang, Q. Nanostructured Conjugated Polymers: Toward High-Performance Organic Electrodes for Rechargeable Batteries. ACS Energy Lett. 2017, 2, 1985–1996.
- Nakahara, K.; Oyaizu, K.; Nishide, H. Electrolyte anion-assisted charge transportation in poly(oxoammonium cation/nitroxyl radical) redox gels. J. Mater. Chem. 2012, 22, 13669–13673.
- Chae, I.S.; Koyano, M.; Oyaizu, K.; Nishide, H. Self-doping inspired zwitterionic pendant design of radical polymers toward a rocking-chair-type organic cathode-active material. J. Mater. Chem. A 2013, 1, 1326–1333.
- Tokue, H.; Murata, T.; Agatsuma, H.; Nishide, H.; Oyaizu, K. Charge–Discharge with Rocking-Chair-Type Li+ Migration Characteristics in a Zwitterionic Radical Copolymer Composed of TEMPO and Trifluoromethanesulfonylimide with Carbonate Electrolytes for a High-Rate Li-Ion Battery. Macromolecules 2017, 50, 1950–1958.
- Suga, T.; Sakata, M.; Aoki, K.; Nishide, H. Synthesis of Pendant Radical- and Ion-Containing Block Copolymers via Ring-Opening Metathesis Polymerization for Organic Resistive Memory. ACS Macro Lett. 2014, 3, 703–707.
- Wang, S.; Li, F.; Easley, A.D.; Lutkenhaus, J.L. Real-time insight into the doping mechanism of redox-active organic radical polymers. Nat. Mater. 2019, 18, 69–75.
- Koshika, K.; Sano, N.; Oyaizu, K.; Nishide, H. An Aqueous, Electrolyte-Type, Rechargeable Device Utilizing a Hydrophilic Radical Polymer-Cathode. Macromol. Chem. Phys. 2009, 210, 1989–1995.
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85.
- Li, F.; Zhang, Y.; Kwon, S.R.; Lutkenhaus, J.L. Electropolymerized Polythiophenes Bearing Pendant Nitroxide Radicals. ACS Macro Lett. 2016, 5, 337–341.
- Xie, Y.; Zhang, K.; Monteiro, M.J.; Jia, Z. Conjugated Nitroxide Radical Polymers: Synthesis and Application in Flexible Energy Storage Devices. ACS Appl. Mater. Interfaces 2019, 11, 7096–7103.
- Liu, K.; Perera, K.; Wang, Z.; Mei, J.; Boudouris, B.W. Impact of open-shell loading on mass transport and doping in conjugated radical polymers. J. Polym. Sci. 2021, 59, 2771–2782.
- Li, F.; Wang, S.; Zhang, Y.; Lutkenhaus, J.L. Electrochemical Energy Storage in Poly(dithienopyrrole) Bearing Pendant Nitroxide Radicals. Chem. Mater. 2018, 30, 5169–5174.