While most biosimilars have rigidly followed the WAC pricing [7] of 3–30% below the reference product in the US, we can anticipate significant price drops in the future. Since the US market represents 40% of the world market [8], price drops in the US are pivotal in presenting the overall benefits of biosimilars. In the EU, the pricing of biosimilars is a regional issue, presenting a range of 30 to 70% of market share and price drops of up to 85%, with Norway, Denmark, and Italy leading the reductions [8]. The success of biosimilars in Europe was due to them achieving widespread acceptance by payers, providers, and patients as an integral part of medicine through an extensive program involving preparing stakeholders, investing in evidence generation (e.g., the NorSwitch trial), and introducing incentive models to share payer savings with hospitals. A key component of this success was forced switching, which is not possible on legal grounds in some countries and due to commercial interest in others.
1. Introduction
While most biosimilars have rigidly followed the WAC pricing [
7] of 3–30% below the reference product in the US, we can anticipate significant price drops in the future. Since the US market represents 40% of the world market [
8], price drops in the US are pivotal in presenting the overall benefits of biosimilars. In the EU, the pricing of biosimilars is a regional issue, presenting a range of 30 to 70% of market share and price drops of up to 85%, with Norway, Denmark, and Italy leading the reductions [
8]. The success of biosimilars in Europe was due to them achieving widespread acceptance by payers, providers, and patients as an integral part of medicine through an extensive program involving preparing stakeholders, investing in evidence generation (e.g., the NorSwitch trial), and introducing incentive models to share payer savings with hospitals. A key component of this success was forced switching, which is not possible on legal grounds in some countries and due to commercial interest in others. For the most popular products, such as adalimumab, erythropoietin, filgrastim, infliximab, rituximab, etanercept, and trastuzumab, almost 100% of the market is held by biosimilars [
8].
The EU has approved 84 products [
9], 70 of which are on the market, compared to 34 approvals in the US [
10], of which 19 are on the market [
11]. The reference products for all of these are only 9 molecules in the US and 17 in the EU; the exceptions in the US include insulin lispro, insulin aspart, follitropin alfa, epoetin zeta, teriparatide, and enoxaparin sodium. Enoxaparin is considered a drug by the FDA and is not reported as a biosimilar [
12]; it was approved as a drug in the EU, but now both the EU and Canada consider it a biological product that has qualified to be approved as a biosimilar. Although beginning in 2020, the FDA moved to regulate many products that were treated as drugs by the FDA, including insulins and hormones, as biologics, teriparatide is not on the FDA’s list of products that will undergo this regulatory transition [
13].
In their September 2020 report, the IQVIA Institute for Human Data Science estimated biosimilar sales to total USD 80 billion over the next five years compared to the USD 14 billion made during the previous five years (2015–2019); they also projected that the availability and use of biosimilar medicines would reduce US drug costs by USD 100 billion by 2024. In their January 2022 report, the IQVIA updated its global estimates, showing projected biosimilar sales of about USD 40 billion in 2025 and USD 75 billion in 2030 [
14].
Biologics represent 34% of pharmaceutical spending in Europe, reaching EUR 78.6 billion in 2021 and growing at a 10.5% compound annual growth rate (CAGR) over the past five years. The total European biosimilar market has reached EUR 8.8 billion in 2021 [
5], accessing between 10–40% of the total biologics market by country. By 2020, the list price savings (excluding confidential rebates and discounts) accounted for EUR 5.7 billion versus the pre-biosimilar cost of the originator. This figure would likely be even higher based on net prices.
At present, there are substantial commercial opportunities for biosimilar companies. However, with the anticipated lowering of the cost in the market, the level of competition is expected to rise, which will eventually benefit patients. Price reductions of 70% to 80% are needed to encourage the adoption of biosimilars globally.
2. The US Scene
There are over 100 biosimilar programs enrolled with the FDA [
33]. To expedite the approval process, the FDA has taken several significant steps.
-
The FDA has created two new guidelines, the extension of the Q&A presentations [
34] and the third revised draft guidance [
35] titled “New and Revised Draft Q&As on Biosimilar Development and the BPCI Act”. The details refer to fulfilling pediatric assessment or PREA requirements, post-approval filing, and the assertion that the 351(k) cannot have a different route or dosage form. However, the strength issue was delayed, adding new indications and orphan exclusivity. The FDA also updated The Purple Book FAQ section [
36].
-
FDA has also published new fact sheets [
37] to provide additional educational materials on biosimilar and interchangeable products and the biosimilar regulatory review and approval process. The BPCIA states [
38] that the “Secretary may determine, in the Secretary’s discretion, that an element described in clause (i) (I) [the biosimilar testing] is unnecessary in an application submitted under this subsection”. The FDA has subtly implemented this change in its new biosimilar guidance [
39].
-
The BPCIA text [
38] states that “an application submitted under this subsection shall include information demonstrating that the biological product is biosimilar to a reference product based upon data derived from analytical studies, animal studies, and clinical studies”. The new education material includes the phrase “in addition to analytical studies, other studies that may be needed”, not shall be, as stated in the BPCIA.
-
Animal studies are now described as unnecessary for providing toxicology or pharmacology information about a biosimilar.
-
Clinical pharmacology studies show that the proposed biosimilar passes through the body the same way as the reference product and has the same effects. This could include an immunogenicity test to see how the biosimilar affects a patient’s immune system.
-
Additional clinical studies can sometimes be conducted after other studies to address any remaining uncertainty about whether the proposed biosimilar has clinically meaningful differences from the reference product.
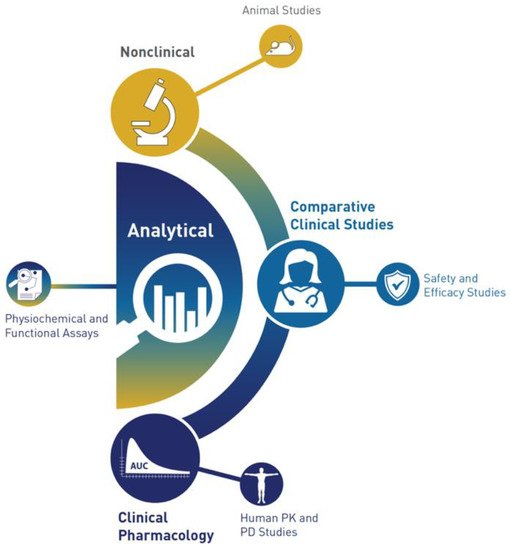
The historic pyramid of the FDA is now replaced with a crescent [
40] (
Figure 2).
3. The European Scene
The EU was the first region to develop a robust regulatory framework for the authorization of biosimilars. In 2001, much of the EU’s directive-based legislation concerning the regulation of medicines was codified as Directive 2001/83/EC [
41]. The European Medicines Agency (EMA) has developed a regulatory scheme for biosimilar authorization through a relatively transparent process. The EMA has issued concept papers and draft guidance and held public scientific workshops. It has issued guidelines that describe general principles and provided an overarching framework for the authorization of biosimilars. The EMA’s Committee for Medicinal Products for Human Use (CHMP) has also issued product class-specific guidance that sets out product requirements in greater detail. For example, the CHMP has issued guidelines for recombinant erythropoietin, granulocyte-colony stimulating factor, recombinant human soluble insulin, low-molecular-weight heparins, somatropin, and recombinant interferon alfa [
42]. EMA has announced that they intend not to issue more specific biosimilar guidelines but instead prefer to give tailored advice on a case-by-case basis [
43].
It is anticipated that the EMA will add more guidelines, particularly for monoclonal antibodies, that will significantly reduce the regulatory barrier and the cost and time taken to reach the market. The FDA has stayed away from creating product-specific guidelines and discouraged the USP from creating any monographs for biological drugs to prevent the originators from including specifications that might be protected intellectual property.
In 2022, the CHMP initiated a new program to engage additional stakeholders during the discussions relating to the evaluation of the development of products; it is anticipated that more changes to the regulatory control of biosimilars will result from this effort [
42]. In addition, the EPAR program of CHMP is an excellent source of information and learning for biosimilar developers; 84 dossiers are available [
44]. The FDA also posts details of its approval of biosimilar products. However, a biosimilar developer may object to the posting, in which case the details can only be secured under the Freedom of Information Act [
45].
This entry is adapted from the peer-reviewed paper 10.3390/biologics2020009