Monoclonal antibody fragments (Fab) are a rising class of biotherapeutics and their production has been optimised using coexpression of molecular chaperones such as DsbC or DnaK–DnaJ–GrpE, as well as strain engineering for post-translational modifications such as disulphide bridging. Different media systems such as EnBase and combining nitrogen source supplementation with low-temperature cultivation have resulted in improvement in cell integrity, protein expression, and protein refolding. The recovery of native proteins from insoluble inclusion bodies can be improved by adjusting refolding conditions, as well as by incorporating multimodal and affinity chromatography for achieving high product yields in purification.
1. Major Developments in Upstream Processing of Fab Fragments in E. coli
Production of a Fab fragment in a bacterial host is an attractive option due to the above-mentioned advantages. Recent advances in cloning technologies and host cell engineering along with process development have resulted in a significant improvement in the expression of Fab molecules in E. coli.
1.1. Expression and Localisation of Fab Fragment
Production of Fab fragments in
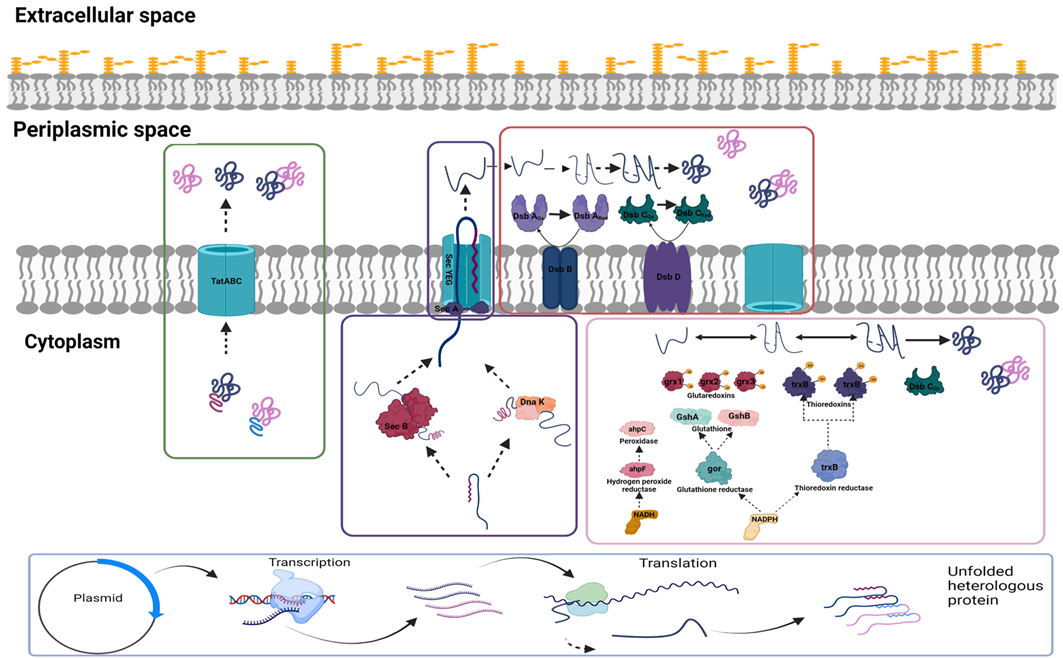
E. coli typically offers multiple challenges, including low expression, poor yield of properly folded Fab fragments, and unequal expression of heavy and light chain in case of bicistronic expression. Oxidising conditions that allow disulphide bond formation are critical for the soluble expression of Fab fragments. This is achieved by directing the expressed molecule to the periplasmic space, which provides the oxidising environment required for disulphide bond formation. Inefficient translocation is one of the major hurdles in periplasmic expression, and different Sec pathways and codon modulation in signal sequences have been tried to increase the expression of properly folded soluble fractions efficiently. The pertinent cytoplasmic and periplasmic pathways have been illustrated in
Figure 1. The PelB signal sequence library was created by codon modulation, and its effect on Fab expression has been reported [
13]. Further, the fifth leucine position of light chain PelB has been proposed to be significant for Fab fragment expression. Fab leakage has been observed in the case of OmpA-signalled Fab, and researchers have used different signal sequences to replace heavy and light chain OmpA [
14]. Further, DsbA has been used to prevent leakage and for re-routing of secretory pathways to rescue the poor performance, with this strategy demonstrating significant improvement in Fab expression. In a recent development, it has been observed that when OmpA of both chains is replaced with DsbA, uncleaved soluble light chains are observed in both cases and the amount of uncleaved soluble fragments is more in the plasmid-based system, compared with genome-integrated clones. Based on these observations, we can conclude that the use of different signal sequences for heavy and light chains can increase the periplasmic translocation, as well as cleavage efficiency, and can prevent overburden on the signal translocation pathway [
15].
Figure 1. Schematic of (A) translocation pathways: Sec- and Tat-dependent protein transport pathways. The Sec pathway transports loosely folded or unfolded proteins across the plasma membrane. Cytoplasmic targeting factors such as SecB help control protein targeting and folding. On the other hand, the SecYEG channel and the translocation ATPase SecA use ATP as a driving force to push the protein through the membrane. The transmembrane proton gradient may also power translocation. Periplasmic protein folding is mediated by oxidases DsbA and DsbC and chaperones, which give final active and protease-resistant conformation of translocated protein. Unlike the Sec pathway, the Tat pathway transports fully folded cofactor proteins. TatA, TatB, and TatC may make up the Tat translocase. Transmembrane proton-motive force drives protein transport via Tat; (B) cytoplasmic disulphide bond formation: Thioredoxin (TrxB) and glutaredoxin (Gor) pathways are required for reducing cytoplasmic protein disulphide bonds. While one relies on glutathione reductase and tripeptide glutathione, the other relies on thioredoxin reductase and one of the two thioredoxin family members. They both use NADPH. Glutaredoxins (Grx1, Grx2, Grx3) and oxidised thioredoxins (TrxB) catalyse the reversible oxidation–reduction of protein disulphide groups. Cytoplasmic DsbC isomerises mis-oxidised proteins to their native correctly folded state. (Image created in BioRender.com).
1.2. Co-Expression of Soluble Expression Partners
Insoluble aggregate formation and coexpression of molecular chaperones with Fab fragments to enhance the solubility and proper folding of antibody fragments are being widely studied as an effective approach to overcome protein loss and reduce process time at the refolding step in the case of inclusion bodies.
The expression of periplasmic chaperones such as DsbC exhibits a synergistic effect on protein folding and disulphide bond formation. In the cytoplasm, coexpression of DnaK–DnaJ–GrpE chaperones facilitates protein folding and transport across the membrane (
Figure 1). DnaK–DnaJ–GrpE chaperones coexpressed with anti-TNF-α Fab in the BL21 host system result in a marked increase in the periplasmic translocation and active soluble expression [
17]. DsbC has a positive effect on the solubility of the proteins expressed in the cytoplasm but not in the proteins targeted to the periplasmic space. In another study, it has been observed that coexpression of the DsbC chaperon in periplasmic protease deficient strains such as wild-type W3110 and MXE001 strain resulted in high periplasmic Fab yield and increased levels of cell viability for four different types of Fab. A twofold yield increase in three Fab molecules (average 1.1 g/L to 2.25 g/L) and a fivefold (average 0.48 g/L to 2.6 g/L) increase in one Fab molecule was observed, as compared with wild type. Additionally, an increase in cell viability up to 40 h post-induction was observed in these modified strains (from around 80 OD to 105 OD) [
18]. Since only a few pathways transfer folded or partially folded proteins to periplasmic space, researchers have examined the effect of coexpression of different sets of cytoplasmic chaperones on the yield of the Fab fragments and observed that the DnaKJE (DnaK–DnaJ–GrpE) had a positive effect on solubilising recombinant proteins expressed in the cytoplasm and had no effect on increasing the functionality of protein [
6,
17]. In the same study, it was reported that GroES–GroEL did not improve solubility and functionality, and it was concluded that GroES–GroEL chaperones are likely more effective in the case of cytoplasmic expression than the periplasmic expression.
1.3. Strain Development and Protein Expression
Proteolytic degradation is the primary concern when expressing heterologous proteins in prokaryotic host systems. Recent advances in strain engineering have enabled us to improve the tolerance and applicability of host cells for the expression of heterologous proteins.
In the case of periplasmic expression of Fab fragments, researchers have developed Tsp, protease III, and DegP periplasmic protease deficient strains, and an increase in protein expression was observed in the Tsp deficient strain. Coexpression of protease deficient strains with DsbC gave increased yield and also restored cell viability; the final yield of 2.4 g/L was achieved, compared with 1 g/L in wild-type strain [
18]. This indicated the likely involvement of Tsp in the degradation of Fab in periplasmic space, which was tested in wild-type W3110 and MXE001 strains and showed consistent results in both cases.
The CyDisCo system, which is based on the coexpression of Erv1p, DsbB, or VKOR, and either DsbC or PDI, has been developed for the production of disulphide bond containing proteins in
E. coli. Researchers have studied the efficiency of the CyDisCo system to express Fab and scFv in
E. coli, and it has been reported that the CyDisCo system is able to generate high yields of folded, biologically active, antibody fragments in the cytoplasm of
E. coli with more than 90% success rate. In one study, four Fabs were used, and a nearly 20-fold increased yield was observed. In addition, 42 mg/L folded purified Fab (Maa48) was obtained from a shake flask study, and overall general average Fab levels were 23 mg/L [
19].
Genomic integration of the gene of interest has been attempted by performing site-directed integration of the gene of interest into the host cell genome [
15]. When compared with the plasmid-based and genomic integration-based systems, the proposed approach resulted in yields ranging from 80% to 300%, whereas plasmid-based counterparts were considered at 100%. Soluble specific Fab yields ranged from 0.01 mg to 7.4 mg Fab per gram of cell dry mass. This study also proved that the genome-based system is a better option for expressing proteins translocated to the periplasm. The genomic integration lowers the concentration of mRNA, which prevents the overburdening of the translation, translocation, and folding machinery. This can also be used as an alternative to a plasmid-free, antibiotic selection pressure-free system.
1.4. Fermentation Process and Media Development
After gene level modification and strain engineering, process development primarily focuses on media components and cultivation parameters. The former entails examining suitable combinations of carbon source, nitrogen source, and complex rich media with different combinations of yeast extract, peptone, and tryptone. Researchers have also optimised different process parameters such as pH, agitation, temperature, cell density, the concentration of inducers, induction time, and OD.
The use of a suitable carbon source and its concentration during different stages of expression is the main factor affecting cell growth and protein expression. Excessive glucose supplementation leads to higher acetate production, which affects cell growth and product yield. The EnBase system, in which glucose is gradually released into the medium by enzymatic degradation of glucose polymer, was applied for the expression of Fab molecules, and it was observed that better protein yield and ratio of soluble to total protein were achieved in
E. coli BL21 [
22]. Higher biomass production and lower acetate accumulation resulted in improved recombinant protein expression. Similar observations have been reported by other researchers, in studies in which the use of the EnBase system resulted in a 3-times increase in biomass, compared with LB in both BL21 and TSHuffle host systems [
1]. While a 19% increase in protein expression was reported in the TSHuffle strain, the difference was minor in the case of the BL21 strain. The highest achieved in this cultivation mode was 12 mg/L.
2. Major Developments in Downstream Processing of Fab Fragments in E. coli
2.1. Refolding
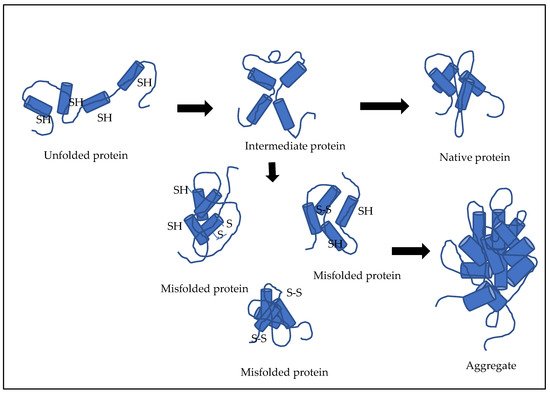
Recombinant antibody fragments such as Fab molecules, when produced in E. coli in the form of misfolded aggregates called inclusion bodies (IBs), need to be refolded into a functionally active protein (Figure 2). Refolding is often a rate-limiting step and a major challenge in the production of such Fab molecules owing to the shuffling of disulphide bonds. These multidomain proteins carry intra- and interchain disulphide bonds, which makes them highly complex molecules, such that the refolding becomes an overall determining step for cost-effective and time-effective production of these biotherapeutics.
Figure 2. Illustration of refolding of multidomain proteins.
There are various events that occur during the refolding of these multidomain proteins. These include domain association, structural changes based on hydrophobicity, bond formation involving hydrogen or electrostatic interaction, and disulphide bond formation [
12]. The association of light and heavy chains via disulphide bridges is highly inefficient in
E. coli due to the large number of combinations that can occur in making and breaking the disulphide bonds [
24,
25]. The refolding efficiency is highly constrained due to the higher propensity of aggregation formation owing to higher-order reaction kinetics than refolding, which is a first-order reaction [
26]. Thus, a deeper understanding of molecular behaviour is required to enhance the refolding of these multidomain proteins. Researchers have studied the in vitro unfolding of Fab molecules by applying two- and three-state thermodynamic models, followed by DoE-based optimisation [
12]. The refolding kinetics was examined, and it was surmised that refolding follows a three-state mechanism, wherein the formation of intermediates by light and heavy chains was the rate-limiting step. For renaturing of proteins, redox agents are often employed, wherein the reduced and oxidised forms are added in an optimum ratio to favour the refolding process. These forms include the glutathione system (GSH/GSSG), cysteine/cysteine system, DTT/GSSG system in the molar ratio range of 1:1 to 10:1 [
27]. To enhance refolding yield, various additives are added to the refolding buffer. These additives can act during the different conformations that the protein acquires during refolding, such as stabilising the native state, increasing the reaction kinetics towards correct folding, or suppressing aggregation during the formation of intermediates. Some of the commonly used additives are sugars, polyethylene glycol (PEG), urea, acetone, dimethyl sulphoxide (DMSO), and amino acids such as arginine [
28,
29].
In hosts other than
E. coli, refolding has also been a limitation in yeast,
Pichia pastoris [
30], and insect cell-based expression systems [
31,
32]. Even though mammalian-based expression systems can produce a refolded biotherapeutic, the cost of production increases considerably. Therefore, there is a need to address the challenges posed by the refolding unit operation for the efficient production of these recombinant biotherapeutic proteins.
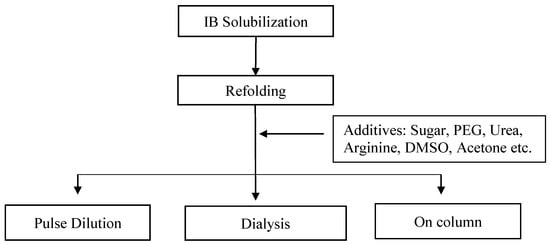
A variety of approaches have been suggested by researchers for the refolding of Fab biotherapeutics. These approaches involve techniques such as drip dilution, dialysis, and on-column refolding (Figure 3).
Figure 3. Different methods of refolding and various additives utilised in refolding.
To enhance the refolding efficiency, the solubilisation and unfolding behaviour of Fab molecules need to be studied. A group of scientists determined the unfolding events using nano-differential scattering fluorimetry (nano-DSF) [
12]. The unfolding of proteins usually follows two-state models, which have native and unfolded forms. In three-state models, there is also an intermediate form along with native and unfolded forms. Understanding these forms can help improve refolding efficiency. In addition, other parameters also determine refolding such as the concentration of chaotrophs, pH of refolding buffer, the temperature during refolding, and the ratio of redox reagents, while the presence of refolding enhancers such as arginine hydrochloride (ArgHCl) determines the overall refolding efficiency. Dilution based in vitro refolding of Fab molecule has been optimised by using the DoE approach, and a refolding yield of 56% has been achieved in 120 h by appropriate dilution in the presence of redox reagents. A recently filed patent covers the process for the production of Fab molecule ranibizumab, in which the refolding of protein was performed by diluting solubilised IBs into refolding buffer, and pH and temperature shift was performed at suitable time intervals to attain the refolded product, with a refolding yield of 13.6% in 22 h [
33].
2.2. Purification of Fab Molecules
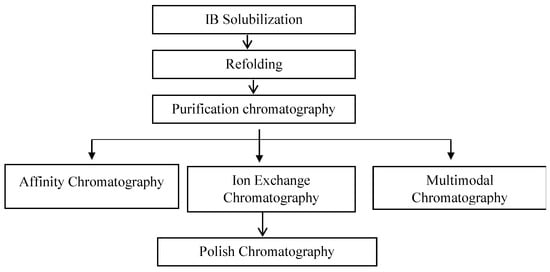
The refolded Fab solution contains a large amount of product-related impurities (residuals of light and heavy chains, intermediates, and aggregates) and process-related impurities (HCPs and HcDNA) that must be removed in order to make a purified drug substance. An extensive study is required for the purification of Fab fragments, as the conventional protein A chromatography performed for mAbs is not applicable due to the lack of an Fc region in Fab molecules (Figure 4).
Figure 4. Conventional process flow chart for the purification of Fab molecules.
Affinity resins such as protein L and protein G are available for the purification of Fab molecules [
38,
39,
40,
41,
42]. Traditional purification schemes involve the use of ion exchange and multimodal chromatography after affinity chromatography [
40,
41,
43,
44]. Various chromatography techniques have been employed by researchers to purify the Fab fragments. The purification of Fab molecules has been demonstrated using two multimodal chromatography resins, and the resulting product had a product purity of 99.50% and an overall process yield of 32.55% [
45]. In one study, researchers performed IgG digestion and isolated Fab fragments using protein A and protein L affinity resins along with ultrafiltration/diafiltration to remove small molecular weight impurities. The overall yield for Fab isolation was approximately 50%. This was followed by resin screening of cation exchange and multimodal resins in the microfluidic device. Their findings suggest that multimodal resins are most suitable the for purification of Fab molecules due to their high salt tolerance and efficient binding of Fab molecules. Furthermore, the microfluidic strategy allows for rapid initial screening of various parameters for designing a purification process. It helps in determining the salt concentration, pH, and adequate resin in a rapid and cheaper way [
40]. Another approach that has been attempted involves the use of cation exchange chromatography (SP Toyopeal resin) to purify the refolded Fab molecule [
36]. An affinity-based apo-B-100-coupled antigen column was successfully used to purify a recombinant Fab molecule specific for apolipoprotein B-100, and a refolded molecule with the production of ~3 mg of purified protein from 1 L
E. coli harvest was generated specific to apolipoprotein B-100, which offered high selectivity for Fab molecule [
37].
This entry is adapted from the peer-reviewed paper 10.3390/fermentation8040175