Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Inorganic & Nuclear
Boron-based bioactive compounds have provided amphiphilic properties to facilitate interaction with protein targets. Indeed, the spectrum of boron-based entities as drug candidates against many diseases has grown tremendously since the first clinically tested boron-based drug, Velcade.
- boron-containing drugs
- boron-containing retinoids
- aminoboronic acids
1. Introduction
Due to its high electrophilic nature, the exogenous element boron and its compounds have been incorporated as key components in various drug candidates, and significant research has been reported over the last decade on the development of new boron-based scaffolds [1,2,3]. Boron has a remarkable ability to create stable electron-deficient three-center, two-electron bonds and strong covalent two-center, two-electron bonds. The covalent ligands present at the boron center can also form strong hydrogen bonds, which can improve effective coordination of active sites in a drug delivery process. In general, based on the literature, these bimodal interactions can be broadly classified into two categories: (i) primary and (ii) secondary interactions [4,5,6].The chemistry is evident from various studies on the behavior of boronic acid/ester present in various drug molecules [7,8,9,10,11,12]. Bortezomib (a dipeptide boronic acid), talabostat (a methanesulfonate salt of L-valinyl-L-boroproline), asborin (carbaborane analogue of aspirin), and tavaborole (a benzoxaborole) represent the remarkable advances in boron chemistry in the field of drug development (Figure 1). Bortezomib (Velcade)was the first boronic-acid-containing drug approved by the Food and Drug Administration (FDA) in 2003. This drug is a proteasome inhibitor that is used to treat multiple myeloma. Because of the overall beneficial properties of boronic acids, since the approval of Velcade, interest in boronic acids has grown in medicinal chemistry, leading to the discovery of two other drugs approved by drug authorities:ixazomib and vaborbactam. Ixazomib, approved by the FDA in 2015, is used to treat multiple myeloma and works via a mechanism similar to that of bortezomib. Vaborbactam, approved by FDA in 2017, is a β-lactamase inhibitor used in combination with antibiotics for the treatment of urinary, abdominal, and lung infections. Benzoxaborole-containing drugs tavaborole and crisaborole, approved by FDA in 2014 and 2017,are used for the treatment of onychomycosis and Eczema, respectively [1,13,14,15,16,17,18].
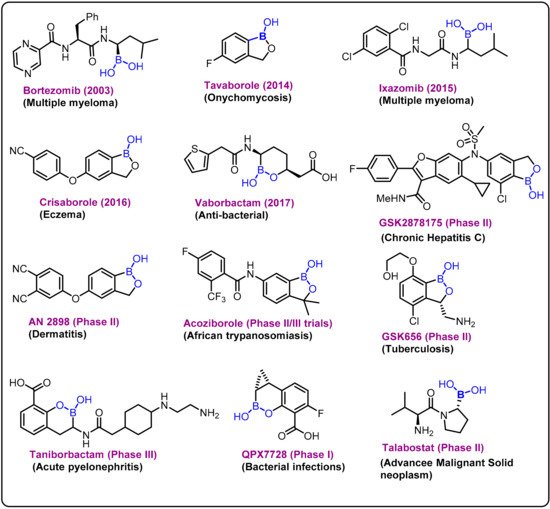
Figure 1. Examples and highlights of boron chemistry in drug discovery.
Mechanistically, the primary interactions involve the boron–hydrogen–boron and Boron–Boron–Boronthree-center, two-electron bonds based on a large compilation of various boron clusters [19,20]. Based on this hypothesis, various carborane derivatives were found to be effective enzyme inhibitors. In particular, metallocarboranes have been found to be effective inhibitors of HIV protease enzymes (Figure 2A). However, the secondary coordinations are extended interactions from the ligands attached to the boron center, as shown in Figure 2B,C. Furthermore, the boron atoms can also be coordinated by strong electron-rich donors, as shown in Figure 2D.
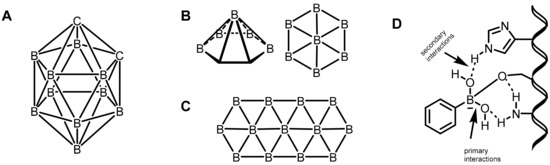
Figure 2. (A) o-carboranes. (B) Borophene layers. Top and side view of a B7 cluster as a constituting unit. (C) Fragment of a B36 borophene molecule. (D) Proposed primary and secondary interactions at the boron center.
One relevant unique feature of the boron atom is its ability to capture neutrons. This has led to several drug discovery platforms. Mechanistically, the nuclear interaction between the boron-10 isotope and a thermal neutron generates an alpha particle, which recoils a lithium-7 atom with a 2.4 MeV. Before traveling one cell diameter, the alpha particle and lithium-ion expend their kinetic energy, resulting in precise cell death. If a compound containing the boron-10 isotope can selectively accumulate in tumor tissue, neutron irradiation can destroy tumor cells without causing substantial damage to normal tissues [21].
2. Boronic Acids/Esters/Aminoboronic Acids as Drug Motifs
One of the most challenging target areas in the field of medicinal chemistry is heart disease. A high level of lipids in the blood (hyperlipidemia) considerably raises the risk of heart disease. This issue has been addressed at the level of transcriptional control for the regulation of lipogenic and cholesterogenic enzymes to maintain effective lipid metabolism. However, progress in treating hyperlipidemia has remained difficult to achieve over the years for two major reasons: (1) the molecular mechanism of lipid metabolism is still unknown and (2) some patients with hyperlipidemia do not respond to statin-related side effects. Therefore, it is necessary to develop novel approaches for the treatment of hyperlipidemia. In this regard, we explored boron-containing stilbene-based compounds and tested them for their potential as lipogenic inhibitors in mammalian hepatocytes. It was found that compound 1 showed a significant decrease in rate-limiting enzymes in the synthesis pathways for both fatty acids and cholesterol at the mRNA level [22]. Interestingly, it was also observed that the particular drug (BF102) has no significant toxicity in mice at the highest possible dose administered in our preliminary in vivo experiments (Scheme 1).
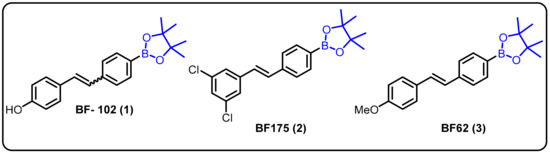
Scheme 1. Boron-containing stilbene derivatives as potential drug candidates.
Dysregulation of lipid homeostasis has become a risk factor, leading to several disorders, such as obesity, hyperlipidemia, insulin resistance, fatty liver, and hypertension [69,70,71]. Among the known lipogenic regulators, sterol regulatory element-binding protein (SREBP) transcription factors are master regulators of lipid homeostasis. The Yang group described boron-based compoundBF175 (2) as a potential lead molecule for the treatment of human diseases caused by dysregulation of lipid homeostasis. BF175 (2) effectively inhibited SREBP target gene expression and lipid biosynthesis in cultured cells by blocking SREBP-TAD binding to MED15-KIX. However, other analogues, such as BF62 (3), failed to show significant activity [23]. In addition, it was the first small-molecule blocker of the physical interaction between MED15-KIX and SREBP-TAD.
A fascinating feature of boronic acids is their ability to act as a serine trap. Hixon et al. studied the influence of various boronic acids against autotoxin [72]. Structurally similar to serine hydrolases, autotoxin is a phospholipase implicated in cardiovascular disease. Autotoxin has a catalytic threonine residue but stabilizes the transient oxyanion through bimetallic coordination and is therefore inhibited by boronic acids. Following the fragment-based screening (FBS) protocol, in comparison to the compounds previously reported by the Kawaguchi group [73], new compounds 4, 5, and 6 were proven to be potent (Scheme 2).
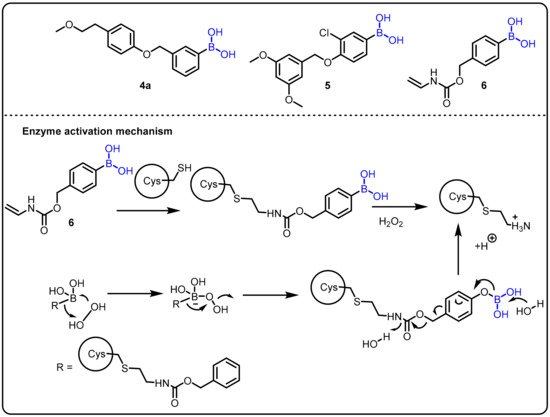
Scheme 2. Boron drug for treatment amyotrophic lateral sclerosis.
Amyotrophic lateral sclerosis (ALS) is an incurable progressive nervous system disease that affects the spinal cord and nerve cells in the brain. It is linked to hyperactive superoxide dismutase (SOD1) and hypoactive angiogenin (ANG), which catalyzes the development of H2O2. Therefore, the chemical reactivity of H2O2, such as the oxidative cleavage of the boron–carbon bond in phenylboronic acid to produce boric acid (B(OH)3) and phenol, could be used in a physiological context. This phenomenon has been used as the basis of chemoselective probes for H2O2 in several cancer prodrug methods. Using this strategy, Hoang et al. introduced a novel synthetic boronic acid mask that restrains the ribonucleolytic activity of ANG [24].Compound 5 accumulates H2O2 and serves to selectively unmask the semisynthetic ANG for the treatment of amyotrophic lateral sclerosis (ALS) (Scheme 2).
Insights into peptide hydrolysis and methods for the development of next-generation proteasome-based cancer therapies can be elucidatedby understanding inhibitory mechanisms. Proteasome inhibition is proposed as an efficient strategy for restricting cancer growth, as well as in the treatment of tumors and hematological malignancies. In this regard, boronic acids have shown an interesting paradox for inhibiting various enzymes at different affected cell sites. Boronic acid inhibitors are particularly noteworthy because bortezomib (7) was the first proteasome inhibitor to be licensed for the treatment of multiple myeloma in 2004 [25]. Insights into the catalytic mechanisms of inhibition necessitate a revised description of the active proteasome site. Ixazomib (8) was approved as an orally bioavailable inhibitor to treat the same disease in 2015, [26] and delanzomib (9) is presently in clinical trials for this purpose and to treat autoimmune disease [27]. In 2016, Chari et al. revealed the crystal structures of the native human 20S proteasome and its complexes with inhibitors [74] (Scheme 3).
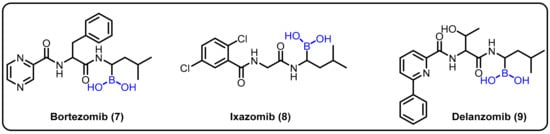
Scheme 3. Boronic-acid-containing proteasome inhibitors.
Various nucleoside analogues, such as 3-thia-2,3-dideoxycytidine (3TC), 3-azido-3′-deoxythymidine (AZT), and 2,3-didehydro-2,3- dideoxythymidine (d4T) have been found to actas antiviral agents targeted at the reverse transcriptase (RT) of HIV [75]. The absence of a 3′ hydroxyl group supportsviral DNA chain termination. Three major resistance mechanisms operate at the RT active sites for viral RT gene mutation to control DNA chain termination under therapeutic conditions; these can be described as: repair of the analogue-terminated DNA, discrimination of the analogue by a decreased affinity (Kd) for RT, and a decreased polymerization rate (kpol) [76]. In this regard, α-boranophosphate nucleosides (α-BH3-dNTPs), as analogues with one non-bridging oxygen on the phosphate substituted by a BH3¯ group, evolved as one efficient strategy to circumvent this resistance [77]. Canard et al. reported BH3-nucleotide analogues as HIV-1 RT inhibitors, having shown the ability to overcome reverse transcriptase-mediated drug resistance [78]. Furthermore, Shaw et al. explained the design and synthesis of cordycepin and 2′-o-methyl α-P-borano triphosphate by using a one-pot phosphorochloridite reaction under mild reaction conditions (Scheme 4) [28,79]. The α-P-borano compounds in which α-phosphateoxygen was replaced by borane were tested in cell culture and displayed potential as effective viral polymerase inhibitors. The overall goal of this project was to improve the antiviral potency of chain-terminating sugar and base-modified purine nucleoside versus the hepatitis C viral RNA-dependent RNA polymerase by using a combination of α-P-borano and nanogel drug delivery technology. Subsequently, a potent thymidylate synthase inhibitor was obtained using a similar boronophosphate scaffold.
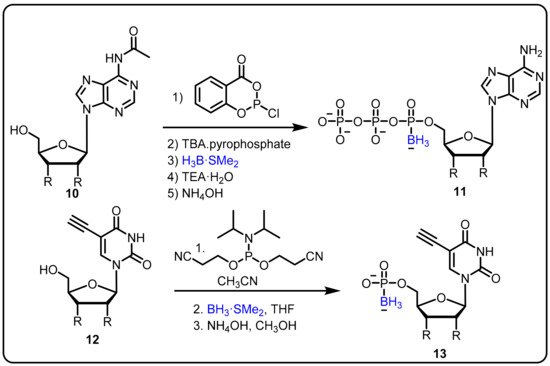
Scheme 4. Boranophosphate nucleosides as an inhibitors of HIV reverse transcriptase (RT).
The Gois group described a novel one-pot approach for the synthesis of boron-containing bioactive scaffolds by the reaction of aryl boronic acids, salicylaldehyde, and amino acids to identify effective enzyme inhibitors. The developed protocol was applied for the discovery of human neutrophil elastase (HNE) inhibitors. Compounds 14 and 15 were found to be the most efficient among the tested substances, with IC50 values of 1.1 and 1.9 μM, respectively, against HNE [29]. Moreover, adetailed mechanistic investigation revealed that imine alkylation and lactone ring-opening are key features ofthe mechanism of inhibition (Scheme 5).
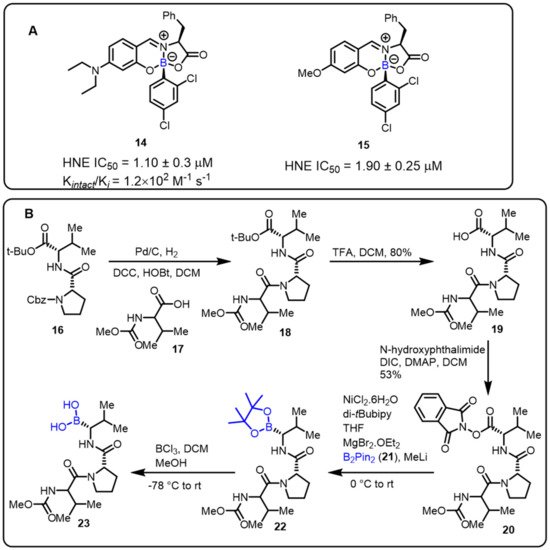
Scheme 5. (A) Borate complexes. (B) Baran’s protocol for the synthesis of HNE inhibitors.
Furthermore, Baran and colleaguesdeveloped an easy decarboxylative-coupling strategy to obtain a highly biologically important boron-containing molecule (Scheme 5) [80]. This method achievedtremendous output in synthesizing Velcade, and one suchhuman neutrophil elastase (HNE) inhibitor compound is now in phase II clinical trials. Amino boronic acids, as potent enzyme inhibitors, can be effectively synthesized in a conventional process via the decarboxylation of the corresponding amino acids.
The anti-TB activity of boromycin (24) against mycobacterial persisters without causing resistance was described by Dick and colleagues. Boromycin was discovered to be active against Gram-positive bacteria and served as a potassium ionophore after being isolated from Streptomy cesantibioticus. Boromycin inhibited mycobacterial growth, with high bactericidal efficacy against both growing and non-growing drug-tolerant persister bacilli (MIC50 = 80 Nm) [30]. Upon exposure to boromycin, intracellular ATP levelsand the membrane potential were reduced, and cytoplasmic proteins were leaked. The addition of KCl to the medium, which was consistent with boromycin working as a potassium ionophore, inhibited its antimycobacterial activity. Boromycin was also shown to have good cytotoxicity and hemolytic activity (CC50 = 30 μM, HC50 = 40 Mm), as well as a selectivity index of more than 300 (Scheme 6).
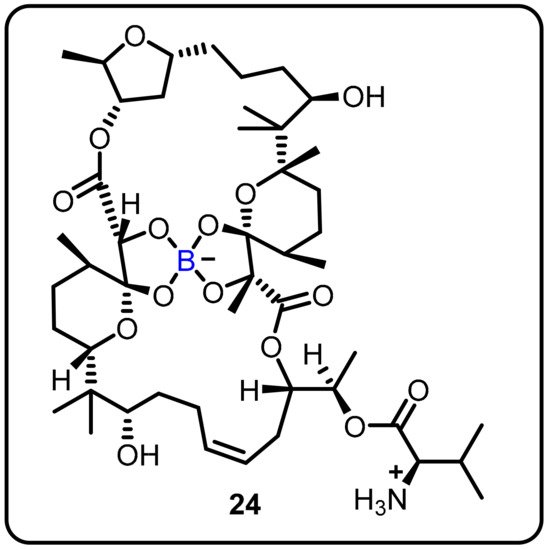
Scheme 6. Boromycin.
Hepatitis C virus (HCV) produces chronic HCV infection in millions of people around the globe, resulting in liver cirrhosis and hepatocellular carcinoma. In this regard, Maynard et al. proposed using boronic acid as a pharmacophore to targeta subset of non-structural protein 5B (NS5B) polymorphs and resistant mutants [33]. The authors designed and synthesized a series of compounds, and among them, compound 25 was found to be an effective potent inhibitor of NS5B polymerase; the authors described boronic-acid-containing moiety as a critical pharmacophore. The potency against key resistant mutants was optimized to afford clinical candidate 25. The most potent compound (25) showed slow binding kinetics with isolated GT1b 316N protein, which was characterized bya division half-life of >40 h (Scheme 7), and also blocked the initial step of the polymerase RNA replication cycle. In hepatitis C virus-infected patients, compound 25 induced a significant reduction in serum HCV ribonucleic acid (RNA) after a single dose of 420 mg (−1.33 log10 copies/mL) compared to the placebo (−0.09 log10 copies/mL) 24 h post-dose (Scheme 7).
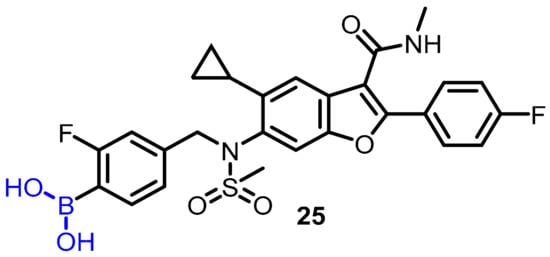
Scheme 7. Potent boronic-acid-derived inhibitor of HCV.
Breakthrough 1
Bortezomib (7) was the first proteasome inhibitor breakthrough approved by the FDA in 2003 for the treatment of newly diagnosed multiple myeloma patients and mantle cell lymphoma [34,35,36,37]. Bortezomib has displayed several clinical benefits, either alone or in combination with other drugs, inducing radio/chemosensitization and overcomingdrug resistance. The overexpression of NOXA is one of the important mechanisms associated with bortezomib’s anticancer activity (NOXA is a proapoptotic protein that interacts with the antiapoptotic Bcl-2 subfamily proteins Bcl-XL and Bcl-2 to cause apoptotic cell death in malignant cells). Another mechanism of bortezomib is suppression of the nuclear factor-κb (NF-κB) signaling pathway, which leads to the downregulation of its antiapoptotic target genes. The majority of bortezomib’s success has been achieved in hematological malignancies, whereas its effect on solid tumors has been less promising. Further significant progress with bortezomib was described by Matsuoka et al. highlighting the growth inhibition of adult T-cell leukemia both in vivo and in vitro [38]. Bortezomib suppressed tumor progression in SCID mice bearing tumors, suggesting that bortezomib has been efficient against adult T-cell leukemia (ATL) cells in vivo. These findings indicate that bortezomib is extremely efficient against ATL cells in vitro and in vivo by induction of apoptosis, and its therapeutic potential could improve the prognosis of patients with this lethal disease.
The Klein group described the boron-based moiety with effective candidates against Zika, West Nile, and Dengue virus proteases. Flaviviruses are responsible for these infections; therefore, inhibition of flavivirus proteases is an interesting approach for the treatment of all three diseases. The authors achieved a thousand-fold affinity by the presence of C-terminal boronic acid moiety into dipeptidic inhibitors, such as protease 32. The desired compound (32) displayed Ki values in the two-digit nanomolar range, and no cytotoxicity was observed, while inhibiting virus replication. In vitro studies revealed corresponding Ki values in the range of Ki = ~0.05, IC50 = 0.066 μM (for Dengue), Ki ~0.08, IC50 = 0.11 μM (for West Nile), Ki ~0.04, IC50 = 0.25 μM (for Zika). (Scheme 8) [40]. Furthermore, the authors explained that boronic acid 32 formed a hydrogen bond with the oxygen in the active site of the flaviviral protease’s Ser135 residue.
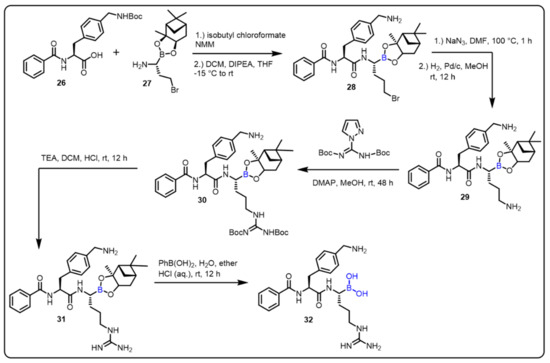
Scheme 8. Peptide-boronic acid inhibitors of flaviviral proteases.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27092615
This entry is offline, you can click here to edit this entry!