The first description of a new form of neutrophil cell death distinct from that of apoptosis or necrosis was discovered in 2004 and coined neutrophil extracellular traps “(NETs)” or “NETosis”. Different stimuli for NET formation, and pathways that drive neutrophils to commit to NETosis have been elucidated in the years that followed. Critical enzymes required for NET formation have been discovered and targeted therapeutically. It has become clear that although this mechanism is thought to enhance host defense by ensnaring bacteria within large webs of DNA to increase bactericidal killing capacity, it is also injurious to innocent bystander tissue. Proteases and enzymes released from NETs injure epithelial and endothelial cells, perpetuating inflammation. In the context of autoimmunity, NETs release over 70 well-known autoantigens. NETs are associated with pathology in multiple diseases: lung diseases, vasculitis, autoimmune kidney diseases, atherosclerosis, rheumatoid arthritis, cancer, and psoriasis. Defining these pathways that drive NET release will provide insight into mechanisms of pathological insult and provide potential therapeutic targets.
- extracellular traps
- inflammation
- neutrophils
- autoimmunity
- cancer
- kidney disease
- arthritis
- lung disease
- COVID-19
- multiple sclerosis
1. Cancer
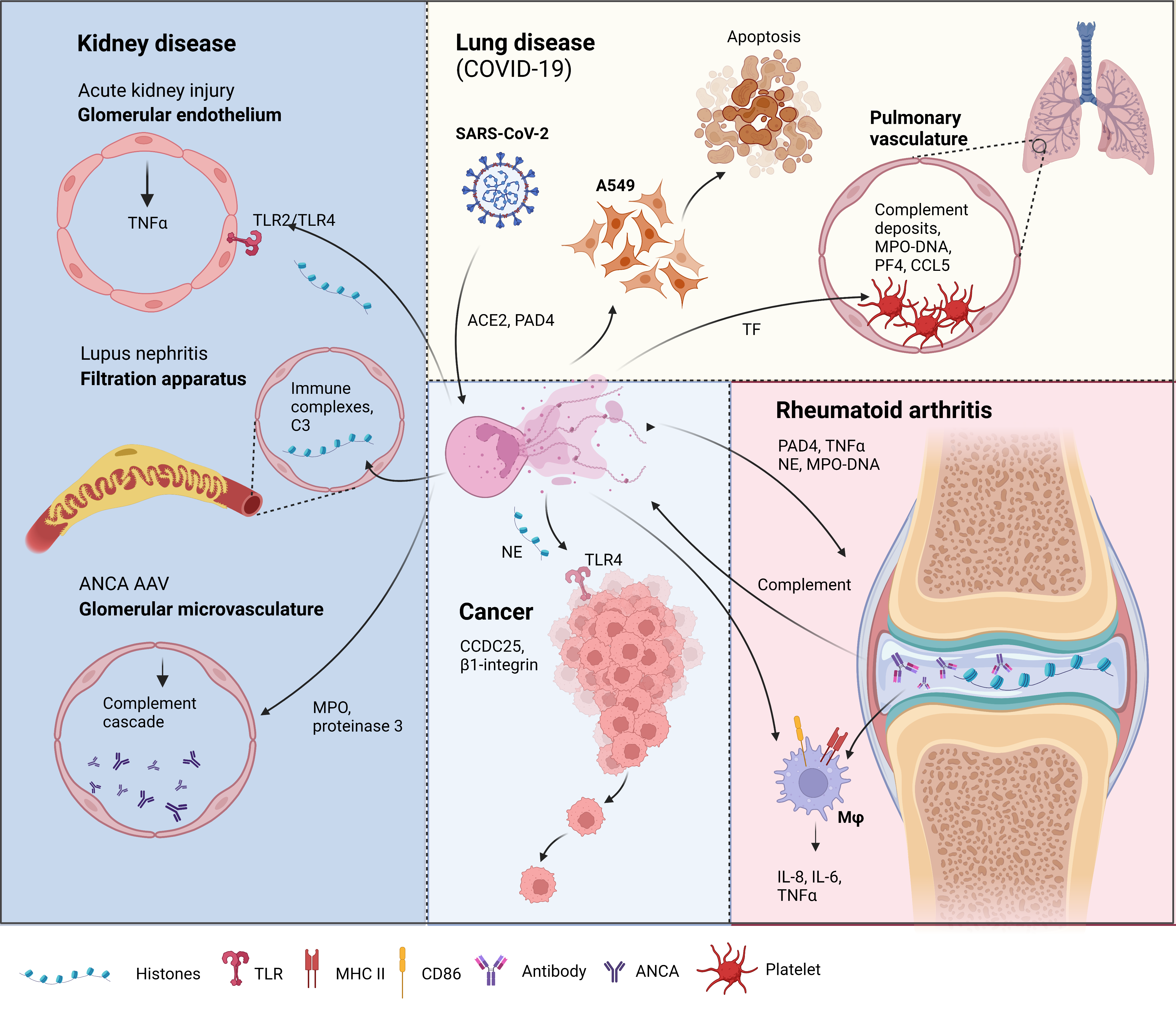
Figure 1. Neutrophil extracellular traps (NETs) in disease pathology. Acute kidney injury: NETosis in the glomerular capillaries results in histone release, triggering renal cell necrosis via Toll-like receptor 2 (TLR2)/TLR4, leading to tumor necrosis factor (TNFα) release [12]. Lupus nephritis: Accumulation of immune complex deposits containing anti-double-stranded DNA (dsDNA) IgG and its target, plus the complement component, C3, attracts neutrophils to the renal filtration apparatus [13]. Presence of hyperacetylated histones in the immune deposits trigger an increase in reactive oxygen species-independent NETosis [14]. Anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV): Neutrophils from AAV patients undergo spontaneous NETosis [15]. ANCA against myeloperoxidase and proteinase 3, two proteins in neutrophil granules are found in the glomerular microvasculature 135 [16]. These autoantigens activate the complement cascade and induce further NETosis [17][18]. Lung disease in coronavirus disease-19 (COVID-19): Severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) can induce NETs via an angiotensin-converting enzyme 2 (ACE2) and PAD4-dependent mechanism [19]. These NETs can enhance the apoptosis of a human alveolar basal epithelial cell line, A549 [20]. Complement deposits in the pulmonary vasculature prime neutrophils for NETosis, resulting in myeloperoxidase (MPO)-DNA complexes. Elevation of platelet factor 4 (PF4), chemokine ligand 5 (CCL5) [21], and the neutrophil–platelet interaction all trigger NETosis [22][23], which was shown to have thrombogenic activity via the upregulation of tissue factor (TF) [22]. Rheumatoid arthritis (RA): Peptidyl arginine deiminase 4 (PAD4) citrullinates histones released by spontaneous NETosis, forming immune complexes in the synovial joint that activate macrophages (mφ) [24]. The immune complexes can also activate the complement cascade, which triggers, further NETosis [25]. NETs from RA donors can activate resting macrophages to induce the surface expression of major histocompatibility complex II (MHCII) and cluster of differentiation 86 (CD86) [26]. These macrophages also release interleukin-6 (IL-6), IL-8, and TNFα [26], which can feedback to induce more NET formation. Cancer: Cancer cells attract neutrophils into the cancer microenvironment (CME), which results in NET formation under the inflammatory CME. These NETs can capture circulating tumor cells via β1-integrin [27], citrullinated histones from NETs can also enhance cancer metastasis by binding to coiled-coil domain-containing protein 25 (CCDC25) [28]. NETs and the neutrophil elastase (NE) released can promote tumor cell growth via TLR4 [29]. Figure created in Biorender.
2. Multiple Sclerosis
3. Kidney Disease
3.1. Acute Kidney Injury (AKI)
3.2. Lupus Nephritis
3.3. ANCA-Associated Vasculitis
3.4. Goodpastures Disease (Anti-Glomerular Basement Membrane Disease)
3.5. Vasculitis
4. Rheumatoid Arthritis
5. Lung Disease
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073793
References
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413.
- Mistry, P.; Kaplan, M.J. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin. Immunol. 2017, 185, 59–73.
- Rother, N.; Pieterse, E.; Lubbers, J.; Hilbrands, L.; Van Der Vlag, J. Acetylated Histones in Apoptotic Microparticles Drive the Formation of Neutrophil Extracellular Traps in Active Lupus Nephritis. Front. Immunol. 2017, 8, 1136.
- Söderberg, D.; Segelmark, M. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis. Front. Immunol. 2016, 7, 256.
- Kitching, A.R.; Anders, H.-J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 2020, 6, 71.
- Radford, D.J.; Savage, C.O.S.; Nash, G.B. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheumatol. 2000, 43, 1337–1345.
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative Complement Pathway in the Pathogenesis of Disease Mediated by Anti-Neutrophil Cytoplasmic Autoantibodies. Am. J. Pathol. 2007, 170, 52–64.
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129.
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153.
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179.
- Stakos, D.; Skendros, P.; Konstantinides, S.; Ritis, K. Traps N’ Clots: NET-Mediated Thrombosis and Related Diseases. Thromb. Haemost. 2020, 120, 373–383.
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414.
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor α through Fcγ receptor IIa engagement by rheumatoid arthritis–specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Care Res. 2008, 58, 678–688.
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Care Res. 2010, 63, 53–62.
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.-C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131.
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 2017, 140, 2321–2330.
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138.
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; van der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019, 79, 5626–5639.
- Mastronardi, F.G.; Wood, D.D.; Mei, J.; Raijmakers, R.; Tseveleki, V.; Dosch, H.-M.; Probert, L.; Casaccia-Bonnefil, P.; Moscarello, M.A. Increased Citrullination of Histone H3 in Multiple Sclerosis Brain and Animal Models of Demyelination: A Role for Tumor Necrosis Factor-Induced Peptidylarginine Deiminase 4 Translocation. J. Neurosci. 2006, 26, 11387–11396.
- Ermert, D.; Zychlinsky, A.; Urban, C. Fungal and Bacterial Killing by Neutrophils. Inflammation 2009, 470, 293–312.
- Naegele, M.; Tillack, K.; Reinhardt, S.; Schippling, S.; Martin, R.; Sospedra, M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J. Neuroimmunol. 2012, 242, 60–71.
- Rumble, J.M.; Huber, A.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35.
- Tillack, K.; Naegele, M.; Haueis, C.; Schippling, S.; Wandinger, K.-P.; Martin, R.; Sospedra, M. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J. Neuroimmunol. 2013, 261, 108–119.
- Paryzhak, S.; Dumych, T.; Mahorivska, I.; Boichuk, M.; Bila, G.; Peshkova, S.; Nehrych, T.; Bilyy, R. Neutrophil-released enzymes can influence composition of circulating immune complexes in multiple sclerosis. Autoimmunity 2018, 51, 297–303.
- Ospina, F.E.; Betancur, J.F.; Suso, J.P.; Muñoz-Buitron, E.; Cañas, C.A.; Tobón, G.J. Role of the cytokine BAFF in autoimmune diseases: Physiopathology and therapeutic targets. Rev. Colomb. Reumatol. (Engl. Ed.) 2016, 23, 177–194.
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A., Jr.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2015, 136, 826–836.
- Yu, G.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. NeuroReport 2018, 29, 208–213.
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron Exp. Nephrol. 2017, 137, 282–286.
- Bolisetty, S.; Agarwal, A. Neutrophils in acute kidney injury: Not neutral any more. Kidney Int. 2009, 75, 674–676.
- Bonavia, A.; Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 2018, 33, 1629–1639.
- Tecklenborg, J.; Clayton, D.; Siebert, S.; Coley, S.M. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150.
- Mulay, S.R.; Linkermann, A.; Anders, H.-J. Necroinflammation in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 27–39.
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768.
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413.
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.-C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131.
- Nawroth, P.; Handley, D.; Matsueda, G.; De Waal, R.; Gerlach, H.; Blohm, D.; Stern, D. Tumor necrosis factor/cachectin-induced intravascular fibrin formation in meth A fibrosarcomas. J. Exp. Med. 1988, 168, 637–647.
- Hertig, A.; Rondeau, E. Role of the coagulation/fibrinolysis system in fibrin-associated glomerular injury. J. Am. Soc. Nephrol. 2004, 15, 844–853.
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45.
- Mistry, P.; Kaplan, M.J. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin. Immunol. 2017, 185, 59–73.
- Kanjanabuch, T.; Kittikowit, W.; Eiam-Ong, S. An update on acute postinfectious glomerulonephritis worldwide. Nat. Rev. Nephrol. 2009, 5, 259–269.
- Tesař, V.; Hruskova, Z. Treatment of proliferative lupus nephritis: A slowly changing landscape. Nat. Rev. Nephrol. 2010, 7, 96–109.
- Menzi, C.P.; Bucher, B.S.; Bianchetti, M.G.; Ardissino, G.; Simonetti, G.D. Management and outcomes of childhood Goodpasture’s disease. Pediatr. Res. 2018, 83, 813–817.
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521.
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J. Immunol. 2011, 187, 538–552.
- Rother, N.; Pieterse, E.; Lubbers, J.; Hilbrands, L.; Van Der Vlag, J. Acetylated Histones in Apoptotic Microparticles Drive the Formation of Neutrophil Extracellular Traps in Active Lupus Nephritis. Front. Immunol. 2017, 8, 1136.
- Zhang, S.; Lu, X.; Shu, X.; Tian, X.; Yang, H.; Yang, W.; Zhang, Y.; Wang, G. Elevated Plasma cfDNA May be Associated with Active Lupus Nephritis and Partially Attributed to Abnormal Regulation of Neutrophil Extracellular Traps (NETs) in Patients with Systemic Lupus Erythematosus. Intern. Med. 2014, 53, 2763–2771.
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818.
- Kallenberg, C.G.M. Pathogenesis of ANCA-associated vasculitides. Ann. Rheum. Dis. 2011, 70 (Suppl. 1), i59–i63.
- O’Sullivan, K.M.; Lo, C.; Summers, S.A.; Elgass, K.D.; McMillan, P.; Longano, A.; Ford, S.L.; Gan, P.Y.; Kerr, P.G.; Kitching, A.R.; et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015, 88, 1030–1046.
- Kitching, A.R.; Anders, H.-J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 2020, 6, 71.
- Radford, D.J.; Savage, C.O.S.; Nash, G.B. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheumatol. 2000, 43, 1337–1345.
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative Complement Pathway in the Pathogenesis of Disease Mediated by Anti-Neutrophil Cytoplasmic Autoantibodies. Am. J. Pathol. 2007, 170, 52–64.
- Yoshida, M.; Yamada, M.; Sudo, Y.; Kojima, T.; Tomiyasu, T.; Yoshikawa, N.; Oda, T.; Yamada, M. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology 2016, 21, 624–629.
- Gadola, S.D.; Gross, W.L. The renaissance of granulomatous inflammation in AAV. Nat. Rev. Rheumatol. 2012, 8, 74–76.
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced Formation and Disordered Regulation of NETs in Myeloperoxidase-ANCA–Associated Microscopic Polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997.
- Söderberg, D.; Segelmark, M. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis. Front. Immunol. 2016, 7, 256.
- Kraaij, T.; Kamerling, S.; Bakker, J.; Brunini, F.; Pusey, C.; Scherer, H.U.; Toes, R.E.; Rabelink, T.; van Kooten, C.; Teng, O. NET-inducing capacity is a biomarker in ANCA-associated vasculitis independent of ANCA antibodies. Arthritis Rheumatol. 2016, 68 (Suppl. 10), 1–15.
- Nakazawa, D.; Marschner, J.A.; Platen, L.; Anders, H.-J. Extracellular traps in kidney disease. Kidney Int. 2018, 94, 1087–1098.
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcγRIIIB but induces neutrophil extracellular traps via FcγRIIA in vivo. Blood 2012, 120, 4421–4431.
- Nishi, H.; Furuhashi, K.; Cullere, X.; Saggu, G.; Miller, M.J.; Chen, Y.; Rosetti, F.; Hamilton, S.L.; Yang, L.; Pittman, S.P.; et al. Neutrophil FcγRIIA promotes IgG-mediated glomerular neutrophil capture via Abl/Src kinases. J. Clin. Investig. 2017, 127, 3810–3826.
- Gordon, R.; Herter, J.M.; Rosetti, F.; Campbell, A.; Nishi, H.; Kashgarian, M.; Bastacky, S.I.; Marinov, A.; Nickerson, K.; Mayadas, T.N.; et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2017, 2, e92926.
- Okazaki, T.; Shinagawa, S.; Mikage, H. Vasculitis syndrome-diagnosis and therapy. J. Gen. Fam. Med. 2017, 18, 72–78.
- Yates, M.; Watts, R. ANCA-associated vasculitis. Clin. Med. 2017, 17, 60–64.
- Yoshida, M.; Sasaki, M.; Sugisaki, K.; Yamaguchi, Y.; Yamada, M. Neutrophil extracellular trap components in fibrinoid necrosis of the kidney with myeloperoxidase-ANCA-associated vasculitis. Clin. Kidney J. 2013, 6, 308–312.
- Nishide, M.; Nojima, S.; Ito, D.; Takamatsu, H.; Koyama, S.; Kang, S.; Kimura, T.; Morimoto, K.; Hosokawa, T.; Hayama, Y.; et al. Semaphorin 4D inhibits neutrophil activation and is involved in the pathogenesis of neutrophil-mediated autoimmune vasculitis. Ann. Rheum. Dis. 2017, 76, 1440–1448.
- Sha, L.-L.; Wang, H.; Wang, C.; Peng, H.-Y.; Chen, M.; Zhao, M.-H. Autophagy is induced by anti-neutrophil cytoplasmic Abs and promotes neutrophil extracellular traps formation. Innate Immun. 2016, 22, 658–665.
- Ishida, H.; Ohkawa, K.; Hosui, A.; Hiramatsu, N.; Kanto, T.; Ueda, K.; Takehara, T.; Hayashi, N. Involvement of p38 signaling pathway in interferon-α-mediated antiviral activity toward hepatitis C virus. Biochem. Biophys. Res. Commun. 2004, 321, 722–727.
- Ma, Y.-H.; Ma, T.-T.; Wang, C.; Wang, H.; Chang, D.-Y.; Chen, M.; Zhao, M.-H. High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res. Ther. 2016, 18, 2.
- Shida, H.; Nakazawa, D.; Tateyama, Y.; Miyoshi, A.; Kusunoki, Y.; Hattanda, F.; Masuda, S.; Tomaru, U.; Kawakami, T.; Atsumi, T.; et al. The Presence of Anti-Lactoferrin Antibodies in a Subgroup of Eosinophilic Granulomatosis with Polyangiitis Patients and Their Possible Contribution to Enhancement of Neutrophil Extracellular Trap Formation. Front. Immunol. 2016, 7, 636.
- Kettritz, R.; Jennette, J.C.; Falk, R.J. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J. Am. Soc. Nephrol. 1997, 8, 386–394.
- Shingu, M.; Nonaka, S.; Nishimukai, H.; Nobunaga, M.; Kitamura, H.; Tomo-Oka, K. Activation of complement in normal serum by hydrogen peroxide and hydrogen peroxide-related oxygen radicals produced by activated neutrophils. Clin. Exp. Immunol. 1992, 90, 72–78.
- Vogt, W. Complement Activation by Myeloperoxidase Products Released from Stimulated Human Polymorphonuclear Leukocytes. Immunobiology 1996, 195, 334–346.
- Wirthmueller, U.; Dewald, B.; Thelen, M.; Schäfer, M.K.; Stover, C.; Whaley, K.; North, J.; Eggleton, P.; Reid, K.B.; Schwaeble, W.J. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J. Immunol. 1997, 158, 4444–4451.
- Dick, J.; Gan, P.-Y.; Ford, S.L.; Odobasic, D.; Alikhan, M.A.; Loosen, S.H.; Hall, P.; Westhorpe, C.L.; Li, A.; Ooi, J.D.; et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental anti-myeloperoxidase glomerulonephritis. Kidney Int. 2017, 93, 615–625.
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767.
- Lundberg, K.; Nijenhuis, S.; Vossenaar, E.R.; Palmblad, K.; Van Venrooij, W.J.; Klareskog, L.; Zendman, A.J.W.; Harris, H.E. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res. Ther. 2005, 7, R458–R467.
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor α through Fcγ receptor IIa engagement by rheumatoid arthritis–specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Care Res. 2008, 58, 678–688.
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Care Res. 2010, 63, 53–62.
- Holers, V.M.; Banda, N.K. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1057.
- Chowdhury, C.S.; Giaglis, S.; Walker, U.A.; Buser, A.; Hahn, S.; Hasler, P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: Analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 2014, 16, R122.
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295.
- Endo, H.; Akahoshi, T.; Takagishi, K.; Kashiwazaki, S.; Matsushima, K. Elevation of interleukin-8 (IL-8) levels in joint fluids of patients with rheumatoid arthritis and the induction by IL-8 of leukocyte infiltration and synovitis in rabbit joints. Lymphokine Cytokine Res. 1991, 10, 245–252.
- Kaneko, S.; Satoh, T.; Chiba, J.; Ju, C.; Inoue, K.; Kagawa, J. Interleukin–6 and interleukin–8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines Cell. Mol. Ther. 2000, 6, 71–79.
- Farrugia, M.; Baron, B. The role of TNF-α in rheumatoid arthritis: A focus on regulatory T cells. J. Clin. Transl. Res. 2016, 2, 84–90.
- Kumar, P.; Banik, S. Pharmacotherapy Options in Rheumatoid Arthritis. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2013, 6, 35–43.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506.
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091.
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129.
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999.
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13.
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; on behalf of the HLH across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034.
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179.
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157.
- Stakos, D.; Skendros, P.; Konstantinides, S.; Ritis, K. Traps N’ Clots: NET-Mediated Thrombosis and Related Diseases. Thromb. Haemost. 2020, 120, 373–383.
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414.
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J. Immunol. 2006, 177, 4794–4802.
- Redecha, P.; Tilley, R.; Tencati, M.; Salmon, J.E.; Kirchhofer, D.; Mackman, N.; Girardi, G. Tissue factor: A link between C5a and neutrophil activation in antiphospholipid antibody–induced fetal injury. Blood 2007, 110, 2423–2431.
- Kourtzelis, I.; Markiewski, M.M.; Doumas, M.; Rafail, S.; Kambas, K.; Mitroulis, I.; Panagoutsos, S.; Passadakis, P.; Vargemezis, V.; Magotti, P.; et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood 2010, 116, 631–639.