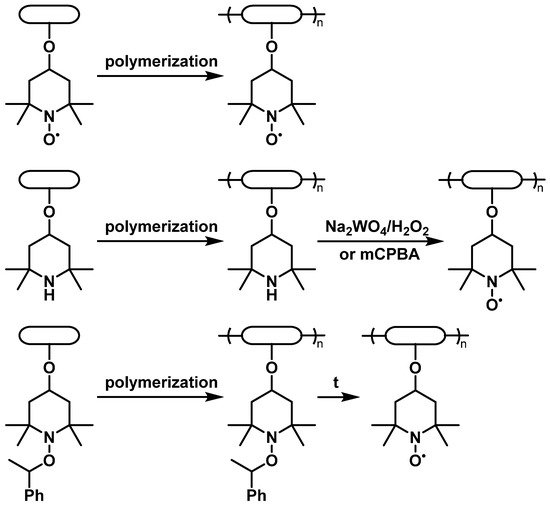
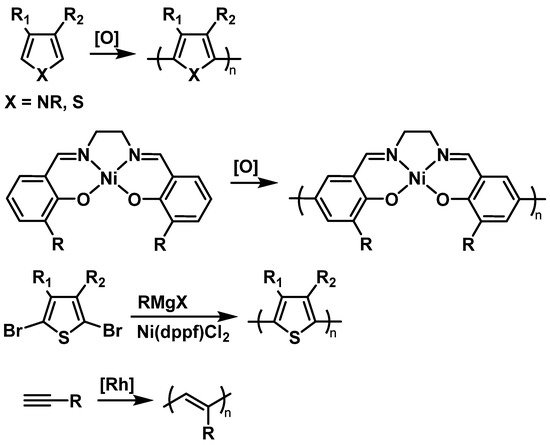
2.2. Fundamental Electrochemistry of TEMPO Transformations
TEMPO is the founding piece among organic radicals for energy storage applications, as well as a touchstone for other similar materials. Its use for lithium-ion batteries can be traced back to 2002 [
43], when Nakahara et al. employed a TEMPO-bearing poly(methacrylate) (PTMA) as an electrode material. Since then, a multitude of polymer backbones has been used in combination with TEMPO and other stable organic radicals [
44], as such materials are quite promising. Their main advantages include high stability in standard lithium-ion electrolytes (recharging with negligible loss up to 2000 cycles [
31]), and fast recharging time (available current rates of up to 100 C [
45]), which results in impressive power density [
11,
12].
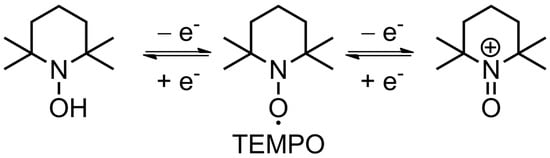
The nitroxide radical can either reversibly oxidize to form an oxoammonium cation (p-doping) or reduce to an aminoxyl anion (n-doping), which is the basis of electrochemical transformations in such materials (
Figure 3). These reactions are useful in a variety of applications, allowing the use of TEMPO-based materials in metal-ion batteries, redox flow cells and oxidation catalysts [
9,
11]. Most reports deal with the oxidation into oxoammonium cation, as this reaction proved to be optimal in terms of its relatively high potential, which is 3.5 V vs. Li/Li
+. TEMPO-modified poly(methacrylate) has become a de facto standard material [
10] for organic cathodes in lithium-ion cells, which theoretical capacity of 111 mAh g
−1 can be achieved in practice. There are other options, such as poly(vinyl ether) with 131 mAh g
−1 [
46], and numerous attempts exist to increase this value up to 224 mAh g
−1 [
47]. However, current materials require binders and conductive agents, practically rendering this value unattainable. Utilization of intrinsically conductive polymers as a backbone might improve the situation. There are also options that allow increase in the cell density via shifting the potential of the reaction up to 3.7 V [
48].
Figure 3. Schematic representation of redox couples of TEMPO radicals.
Regardless, with the average voltage vs. Li being 3.5 V, long cycle life (up to 2000 cycles [
31]) and high (up to 100 C [
45]) rate capability, TEMPO is an ideal candidate for use in organic radical batteries, with power density rivaling that of standard inorganic materials.
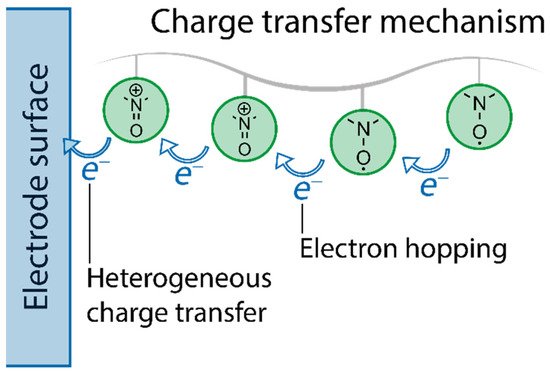
As TEMPO is overwhelmingly used as a pendant group on a polymer backbone, it is reasonable to discuss its electron transfer and doping processes in this context of the whole material, rather than individual TEMPO units. As shown in several studies [
49,
50], electron transfer mechanism includes two distinct steps. The first one is electron transfer between the substrate and the radical components—a heterogeneous step—and the second is self-exchange between the neighboring radicals, primarily driven by concentration gradients of the moieties (
Figure 4). The latter is a homogeneous step and referred to as hopping.
Figure 4. Schematic illustration of the redox reaction in TEMPO-containing polymer between nitroxide radical and oxoammonium cation.
The rate constant of the heterogeneous charge transport is ca. 10
−1 cm s
−1 both for TEMPO monomers [
51], and for TEMPO in PTMA deposited on Pt surface [
52]. The self-exchange between neighboring radicals responsible for the homogeneous transport has a bimolecular rate constant of 1.8 × 10
5 M
−1 s
−1 [
50]. This value is up to four orders of magnitude higher for nitroxide-containing monomers [
51,
53,
54], indicating that attachment of the molecule to the polymer chain hinders diffusion. Still, the fast kinetics cause a small gap between charge and discharge voltages, typically, ca. 100 mV, and allow extraction of almost full capacity of the material at a relatively high 1 C discharge rate: 110 mAh g
−1 actual capacity vs. 111 mAh g
−1 theoretical one [
22].
For the polymer with 100% of monomer units modified by TEMPO (i.e., PTMA), little to no conformational changes accompany electron transfer. This enables Nernstian adsorbate-like behavior in polymer layers with thickness from 10 nm to 100 nm, which allows the use of thicker films and increase in the content of the polymer in the electrode material [
50]. Conversely, the polymer with low density of TEMPO radicals becomes twice as stiff upon oxidation, compared to its reduced state [
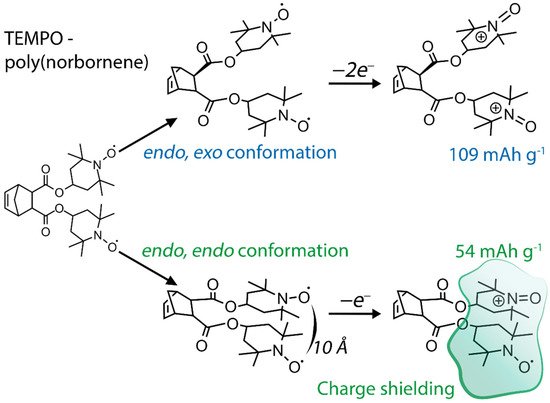
55]. While the electrochemical processes affect the conformation of the polymer, the monomer structure exerts a significant effect on the performance of the materials, such as their capacity. For example, another popular polymer backbone, poly(norbornene), can be functionalized by two TEMPO moieties per monomer unit. In the case of endo/endo configuration of the modified polymer, the capacity of the material is only 54 mAh g
−1, while endo/exo configuration yields full capacity of 109 mAh g
−1 [
56]. Due to the proximity of neighboring TEMPO substituents, which in the endo/endo case are only 10 Å close, the generated positive charge on the first radical prevents oxidation of the second one (
Figure 5). This presents the general issue for implementation of organic radical materials with densely packed redox-active groups.
Figure 5. Schematic illustration of influence of neighboring TEMPO groups on molecule oxidation.
Another aspect worth considering is the intramolecular interactions for TEMPO substituents in long polymer chains, which require increased local concentration of charge carriers to make electron hopping more probable [
57]. One such option is annealing of a polymer with a TEMPO substituent, which results in formation of percolating networks, increasing the conductivity of the polymer to 10 S m
−1, compared to the 1 × 10
−9 S m
−1 of the initial disordered polymer with randomly oriented radical groups [
58].
High electron exchange rates notwithstanding, decent electrode materials performance also involves facile ionic transport. This is directly related to the following requirements to the electrode–electrolyte interactions: the polymer should swell, yet be insoluble, to prevent self-discharging of the electrode [
59], and the polymer should be either solvophilic or well-solvated to provide the mobility of the counterions.
The dissolution of the polymer may result in, e.g., 38% self-discharge in one week [
22], yet this might be easily prevented by crosslinking [
18] of the polymers.
Ensuring ionic conductivity requires tight control over both radical polymer and electrolyte properties. There are multiple factors at play here [
4,
44,
59]. The effect of the porosity of the material matters most in the case of thin-film electrodes, where the increase in thickness would hinder diffusion through the material, as bulk transport becomes the limiting factor [
60], which is observed already from the film thickness of 0.26 μm [
61]. The pores design may be improved via porous carbon architectures [
62], inclusion of porous polymers [
63], or development of three-dimensional electrode structures [
64].
To ensure electroneutrality, the process of electrochemical oxidation of NO• to N
+=O must be accompanied either by anion uptake or by cation expulsion, with the opposite process on reduction. This means that proper selection of material and electrolyte compositions should provide the conditions for high diffusion rates of charged species. Even for typical lithium-ion electrolyte, LiPF
6 (in propylene carbonate, PC), the ratio between the number of TEMPO groups in PTMA and LiPF
6 molecules strongly affects the performance of the material: changing the LiPF
6-to-TEMPO ratio from 1.7 to 0.5 the self-exchange rate constant increases from 0.16 × 10
6 M
−1 s
−1 to 2.8 × 10
6 M
−1 s
−1 [
65]. The decrease in the rate constant in highly concentrated electrolyte has to do with decreased mobility of ions—and predominantly with the nature of anions [
66,
67,
68]—in swollen polymers. It is worth noting, however, that cations also affect the performance of organic radical polymers, e.g., PTMA, and even participate in charge compensation, as shown by electrochemical quartz crystal microbalance [
69].
The replacement of the backbone polymer altogether may enhance ionic conductivity. For instance, (poly(2,2,6,6-tetramethylpiperidinyl-
N-oxyl vinyl ether)) PTVE is hydrophilic, thus providing high affinity to aqueous electrolytes, and ensuring facile ion transport [
70]. Another example is (poly(2,2,6,6-tetramethylpiperidinyl-
N-oxyl glycidyl ether)) PTGE, which has good swelling and ionic transport in standard 1.0 M LiPF
6 in ethylene carbonate: diethyl carbonate (EC:DEC) electrolyte, providing up to 80 mAh g
−1 capacity at 10 C [
27].
As a side note, interactions of TEMPO within the battery may bear disruptive effects on other components by virtue of strong oxidizing capability of TEMPO cation. For example, cellulose-based separators are likely to decompose in its presence [
71]. This presents an additional challenge when selecting optimal components.
Another way to improve the transport properties is to provide doping mechanisms via backbone polymer itself. This can be achieved by using conjugated polymers [
72,
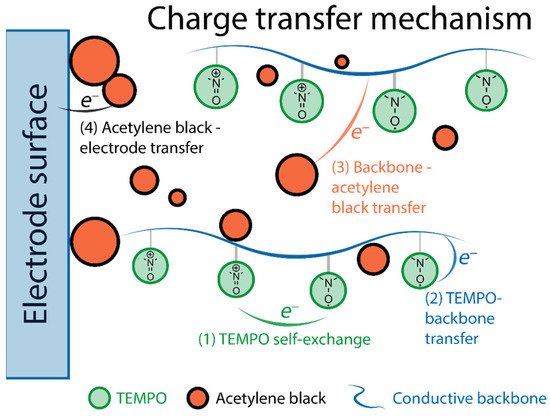
73], though the competing redox processes in radical and backbone components complicate the studies of the charge transport mechanism. Still, there is some understanding of such processes. For example, in TEMPO-modified polypyrrole (with acetylene black as conductive component) [
37] the charge transfer process is separated in four steps: (1) self-exchange between nitroxide radicals, (2) electron transfer from the radical to the conductive backbone, (3) electric conduction from polypyrrole to acetylene black, (4) electron transfer from acetylene black to the current collector (
Figure 6).
Figure 6. Schematic illustration of the charge transfer process in TEMPO-modified polypyrrole.
Polypyrrole is deemed to be an electric conduction path that assists repeatable oxidation/reduction through aggregated radical polymer bulks. The internal charge transfer between the TEMPO pendant groups and the backbone has also been observed in the case of TEMPO-modified 3,4-propylenedioxythiophene (ProDOT) [
74], though the precise effect of such interaction was not firmly established.
Adverse effects of TEMPO radicals have also been reported. In the case of TEMPO-modified poly(3-hexylthiophene) (P3HT) [
35], it turns out that intrinsic conductivity of P3HT—controlled both by intrachain and interchain hopping—is hindered by introduction of TEMPO. The impairing effect of TEMPO is due to introduced disorder in crystalline domains of the polymer, which restricts interchain hopping, thus terminating additional paths of charge transfer and decreasing conductivity. The effects of TEMPO content in P3HT on capacity were also investigated [
48], and, similarly, the capacity was the highest with the minimal content of TEMPO, while 100% TEMPO-loaded conjugated polymer provided both the lowest specific capacity and the highest charge transfer resistance. Another effect was observed for polythiophenes bearing pendant TEMPO groups [
39], where internal transfer of an electron from TEMPO to the polythiophene backbone caused rapid dedoping of the latter. This phenomenon led to a decrease in both capacity and conductivity and due to the thermodynamically favorable conditions: the 3.88 V oxidation potential of polythiophene is higher than that of nitroxide radical (3.60 V), thus facilitating backbone reduction. A reverse scenario, albeit with similar negative effects, was observed for poly(TEMPO−DTP) (TEMPO-modified (dithieno[3,2-b:2′,3′-d]pyrrole)) [
75]: the low oxidation potential of poly(DTP) (3.15 V) caused electron transfer from the backbone to the radical upon charging, thus reducing the activity of the polymer and decreasing Coulombic efficiency. This means that the balance must be found in the thermodynamic preference of the reactions, so that the potentials of both charge carriers overlap in such systems.