Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
The ataxia-telangiectasia mutated (ATM) protein kinase is, as the name implies, mutated in the human genetic disorder ataxia-telangiectasia (A-T). This protein has its “finger in many pies”, being responsible for the phosphorylation of many thousands of proteins in different signaling pathways in its role in protecting the cell against a variety of different forms of stress that threaten to perturb cellular homeostasis.
- ataxia-telangiectasia
- ataxia-telangiectasia mutated protein kinase
- metabolic stress
1. Introduction
Ataxia-telangiectasia (A-T) is an autosomal recessive disorder with features that include immunodeficiency, lung disease, radiosensitivity, cancer susceptibility and neurodegeneration [1,2]. Mutations in the Ataxia-telangiectasia mutated (ATM) gene that codes for a phosphoinositide-3-kinase-like kinase, ATM, are responsible for A-T [3]. The best described stimulus for ATM activation is the presence of DNA damage, primarily DNA DSB [4]. This agrees well with the hypersensitivity of ATM-deficient cells to ionizing radiation, a hallmark of A-T [5,6]. The activation of ATM by DNA DSBs leads to the phosphorylation of thousands of substrates that participate in the control of the cell cycle, DNA DSB repair, apoptosis, transcription and other cellular processes [7]. It seems likely that the alteration to the super-helical structure of chromatin by the introduction of a DSB in DNA leads to a low level of activation of ATM, followed by its recruitment to the sites of the DSB by the MRE11/Rad50/NBS1(MRN) complex and consequent amplification of the activation [8,9]. Autophosphorylation at several sites plays a key role in this process leading to the conversion of an inactive dimer into an active monomer [10]. Once activated, ATM phosphorylates all three members of the MRN complex, which, in turn, mediate the phosphorylation of multiple downstream substrates [11] (Figure 1).
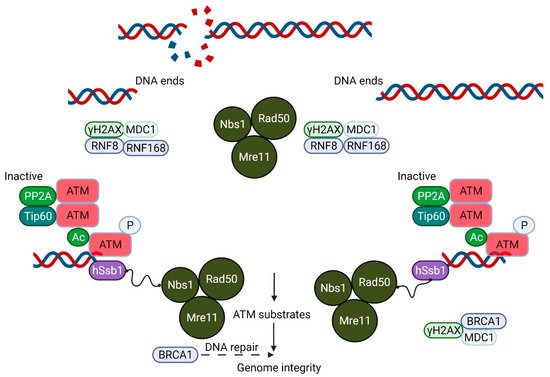
Figure 1. A Schematic model showing the essential role of ATM in DNA double-strand break (DSB) repair. The Mre11/Rad50/Nbs1 (MRN) complex plays an important role in DSB recognition and signaling ATM. ATM, which exists as an inactive dimer under normal conditions, becomes activated as a monomer in response to MRN signaling. This MRN-mediated activation of ATM stimulates the kinase activity of ATM, which phosphorylates p53, Chk2 and other substrates, including Brca1, which together play a crucial role in the maintenance of the genomic integrity of the cell.
ATM is also activated by oxidative and other forms of stress by a distinct mechanism that involves the formation of a disulfide cross-linked dimer [12]. The autophosphorylation of ATM on Ser 1981 and the phosphorylation of its substrates, p53 and Chk2 (not stably associated with DNA), occur in response to H2O2, but the phosphorylation of H2AX, a marker of DNA DSB and Kap1, a heterochromatin protein, both associated more strongly with chromatin, is not observed after H2O2, indicating that this response occurs in the absence of DNA damage (see Figure 2).
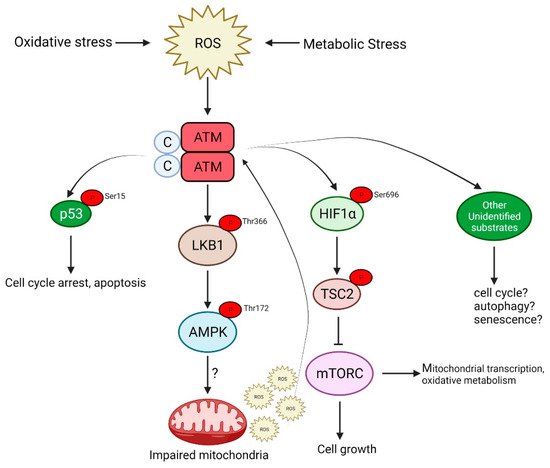
Figure 2. ATM is activated in response to oxidative stress caused by the increased production of reactive oxygen species (ROS). This activation is independent of the DNA damage signaling pathway and signals via AMPK, p53 or mTORC, which may culminate in either of the following: cell cycle arrest, autophagy and senescence or even apoptosis in the case of severe DNA damage.
2. ATM and Inflammation
Early studies on the pivotal role of nitric oxide in the induction of cellular stress and the activation of a p53 response pathway, implicated ATM activation during chronic inflammation [40]. ATM is activated by ROS, which is also elevated in ATM-deficient cells and produces inflammatory changes. A connection between inflammatory stimuli that trigger the activation of the NF-kB response linking DNA damage response to this response, mediated by ATM, has been described [41] (McCool and Miyamoto 2012). The activation of NF-kB regulates the expression of genes involved in the response to oxidative stress and inflammatory changes, and thus, the loss of ATM might be expected to have an impact on this expression. Indeed, genotoxic stress leads to the ATM-dependent phosphorylation of p65(RelA) that represses the transcription of specific genes [42].
There is evidence that systemic inflammation and oxidative stress contribute to the pathophysiology of lung disease in A-T [43]. Serum levels of the pro-inflammatory cytokines, IL-8 and IL-6, were shown to be elevated in patients with A-T, suggesting that markers of systemic inflammation may be useful in identifying the individuals with A-T at an increased risk of lower lung functions [43]. Airway epithelial cells from patients with A-T are also characterized by elevated levels of the pro-inflammatory cytokines IL-8 and TNF-α, following infection with S. pneumoniae, which would also support the role of inflammation in the process. Other evidence for an inflammatory phenotype in A-T patients was provided by Hartlova et al. (2015), who showed that unrepaired DNA damage released into the cytoplasm in Atm−/− mice primed the type 1 interferon response through the STING pathway to promote anti-microbial immunity [44]. The use of olfactory neurosphere-derived cells and brain organoids also revealed that increased cGAS and STING activity was an important contributor to chronic inflammation in the central nervous system in A-T [45]. It was subsequently showed that in the absence of ATM, the defect was in inflammasome-dependent anti-bacterial innate immunity. Diminished interleukin-1B (IL-1B) production, in response to bacteria, was observed in patients and Atm−/− mice. Their data suggested the negative regulation of inflammasome formation by ROS, which could account for the susceptibility of patients to pulmonary bacterial infection. This is keeping with the broader observation that ROS is elevated in all A-T cell types investigated to date. Thus, in addition to the potential of increased ROS to cause DNA and more general cellular damage, these results point to an additional mechanism of interfering with the immune response, and thus increased susceptibility to infection. A role for oxidative damage in impaired innate immunity was also observed in airway epithelial cells from A-T patients [43]. They demonstrated that A-T epithelial cells were extremely sensitive to oxidative stress induced by H2O2, and hypothesized that the increased susceptibility of patients to respiratory infection could be explained by the ability of microorganisms, such as Streptococcus pneumoniae, to produce H2O2 as the damaging agent. They showed that a heightened susceptibility to infection could be explained by both increased oxidative damage and a defect in inflammasome activation. A defect in the ASC–caspase 1 signaling pathway and decreased levels of the inflammasome-dependent IL-1B were observed in patient cells.
3. Mitochondrial Dysfunction in A-T
A mitochondrial disease database was established, which outlines the clinical features observed in these mitochondrial diseases (www.mitodb.com; accessed on 11 February 2022) and allows for the comparison of symptoms in other disorders to determine whether they may fall into a mitochondrial phenotype. Based on this database [46], extensive bioinformatic tools were developed, which revealed that A-T could be characterized as a mitochondrial disorder [46]. The scoring system over a range 0–100 placed A-T at 90 when neurological involvement was included. They suggested that disorders of this nature would thus be prime targets for further investigation, and potentially for treatment with drugs known to augment mitochondrial function. In keeping with this prediction, it is not surprising that there exists an accumulating body of evidence for mitochondrial dysfunction in A-T [47]. The state of continuous oxidative stress and the constitutive activation of pathways that normally respond to oxidative damage, led Gatti and his colleagues to determine whether the oxidative stress phenotype of A-T cells might reflect an intrinsic mitochondrial dysfunction [48]. They observed a sub-population of mitochondria with lower membrane potential in A-T cells, the basal expression of mitochondrial DNA repair and ROS scavenging genes were elevated, mitochondrial respiration and oxidation rates were greatly compromised, and the latter was partially corrected in response to ATM restoration in A-T cells. They concluded that at least some of the oxidative stress observed in A-T cells could be attributed to intrinsic mitochondrial dysfunction. Intrinsic mitochondrial abnormalities in Atm−/− mouse thymocytes were later reported by Valentin-Vega et al., including elevated reactive oxygen species, increased mitochondria mass, a high cellular respiratory capacity, and decreased mitophagy [29]. It was subsequently shown that spermidine triggers PINK1/Parkin-mediated mitophagy in control cells, but not in ATM-deficient cells [49]. Studies in C. elegans and Atm-deficient mice showed that the loss of ATM induced the accumulation of damaged mitochondria, mitochondrial dysfunction and compromised mitophagy due to NAD+ insufficiency [50]. They showed that replenishing intracellular NAD+ improved mitochondrial quality by increasing mitophagy. Furthermore, it reduced the severity of neuropathology and extended the lifespan of both animal models. The conclusion from these data was that both the accumulation of DNA damage and mitochondrial dysfunction contribute to the pathophysiology of premature aging in A-T.
The role of ATM in more general autophagy is somewhat confusing in that it has been reported to be impaired, and both upregulated and downregulated in ATM-deficient cells [29,51,52]. For example, it has been reported that autophagic flux is upregulated in Atm−/− lysosomes associated with a more acidic pH. In short, when ATM is missing or inhibited, autophagy levels increase, which appears to be the opposite to the defect in mitophagy in several reports (see below). Valentin-Vega et al. presented some evidence that autophagy was upregulated in ATM-null thymocytes and MEFs, showing a marked reduction in p62/sequestosome and an increased ratio of LC3II/LC3I [29]. However, this is evidence for a defect in mitophagy, with striking increases in the number of altered mitochondria and the mitochondrial mass in Atm−/− mouse thymocytes [29]. It is notable that the mitochondrial DNA content varied in different ATM-deficient cells and was noticeably increased only in early passage fibroblasts. It is not clear why this is the case, since increased mitochondrial content would suggest increased mtDNA. The increased mitochondrial content did not appear to be as a result of increased biogenesis, but rather decreased mitophagy. However, the evidence for decreased mitophagy was not clear-cut from Parkin levels, higher basal levels or the lack of change as a result of CCCP treatment, but it was evident that the loss of COX IV was deficient in A-T fibroblasts. It was subsequently shown that spermidine triggers PINK1/Parkin-mediated mitophagy in control cells, but not in ATM-deficient cells [48]. In addition, the formation of mitophagosomes and mitolysosomes, as well as a decrease in mitochondrial mass, were shown to be ATM dependent. Studies in C. elegans and Atm-deficient mice showed a loss of the ATM-induced accumulation of damaged mitochondria, mitochondrial dysfunction and compromised mitophagy due to NAD+ insufficiency [49]. They showed that replenishing intracellular NAD+ improved mitochondrial quality by increasing mitophagy [50]. This was achieved using a mitochondrial reporter strain to visualize the expression and colocalization of LGG-1 and DCT-1 in Atm worms. Furthermore, it reduced the severity of neuropathology and extended the lifespan of both animal models [51]. A significant increase in mitochondrial mass and mitophagy at 50% of that in the controls was more recently observed in ATM-deficient human bronchial epithelial cells after nutrient deprivation [20]. A defect in mitophagy and the increased mitochondrial content reported for A-T cells would be expected to create an imbalance in the copy number and the persistence of damaged mitochondria that would interfere with normal mitochondrial function. However, in a recent report, ionophore-induced mitochondrial damage and mitophagy triggered ATM activation through the generation of the ROS function [52]. While antioxidants inhibited ROS production and ATM activation, but failed to prevent mitophagy, this suggested this form of mitophagy does not require ATM [53]. Indeed, the same group showed that FCCP-induced mitophagy in cancer cell lines was dependent on ATM, but not on its kinase, activity [54]. This would also have an impact on the mtDNA copy number and perhaps its integrity, suggesting a possible role for ATM in the response to DNA damage in mitochondria. The elevated mitochondrial ROS, which appears to be a characteristic of all A-T cell types, would be expected to damage mtDNA [47]. Indeed, there is evidence for a four-fold increase in mtDNA damage in A-T fibroblasts and an impaired capacity to remove oxidative lesions [55]. However, there was some variability between the control fibroblasts in the total lesions and extent of repair. The impaired capacity can be explained by reduced levels of ligase III in mitochondria, in the absence of ATM. This is the only DNA ligase in mitochondria and is required to maintain the integrity of mtDNA. In its absence in mice, severe ataxia is observed [56]. This is the only report of a defect in mitochondrial DNA damage and its repair and needs to be viewed with some caution.
Since Valentin-Vega et al. provided evidence that autophagy was upregulated in ATM-null cells by the allelic loss of the autophagic regulatory gene Beclin-1, they investigated its effect on tumorigenesis [29]. While the development of T-cell lymphomas and Beclin-1 heterozygosity in Atm−/− mice is also associated with tumor development, Valentin-Vega et al. unexpectedly observed that the allelic loss of Beclin-1 in Atm−/− mice increased survival to 262 days compared to 137 days in Atm−/− mice. Furthermore, the delayed tumor development in Atm-null mice associated with Beclin-1 heterozygosity that was correlated with the rescue of mitochondrial defects, not the rescue of DDR abnormalities. However, they could not exclude the possibility that Beclin-1 heterozygosity might also delay tumor development by compromising the survival of developing malignant cells. Nevertheless, Beclin-1 heterozygosity appears to modulate mitochondrial homeostasis, which has an impact on the lifespan of Atm−/− mice. A greater insight into the ATM–Beclin 1 interaction was provided in the demonstration that an ATM/Chk2/Beclin-1 axis protected cells from oxidative stress by promoting autophagy [56]. They demonstrated that Chk2 mediated autophagy by the phosphorylation of Beclin-1, by limiting ROS levels during nutrient deprivation. Under these conditions, by sensing ROS, the ATM/Chk2/Beclin-1 complex would control the levels of ROS, clear damaged mitochondria and prevent cell death. The various abnormalities in mitochondria led Valentin-Vega et al., [27] to suggest that A-T should be considered, at least in part, as a mitochondrial disease.
Since oxidative stress and mitochondrial dysfunction are implicated in A-T [47], it was discussed whether reducing mitochondrial reactive oxygen species (ROS) by overexpressing catalase targeted to mitochondria (mCAT) might alleviate the A-T-related pathology in Atm−/− mice [57]. This approach led to a number of beneficial effects in Atm−/− mice, including a reduced propensity to develop thymic lymphomas, improved bone marrow hematopoiesis and macrophage differentiation in vitro, and the partial rescue of memory T-cell developmental defects. These data support a role for mitochondrial ROS in A-T-related pathology, and the authors raise the possibility that antioxidant therapies directed at mitochondrial ROS may be of therapeutic value for A-T patients. This approach has the benefit, over the more generalized use of antioxidants, of alleviating the A-T phenotype; however, evidently, being able to deliver a construct expressing CAT in the appropriate region or cells in the brains of patients represents a major hurdle. However, what emerges from this report is that being able to improve mitochondrial function by reducing the toxic effects of ROS has the potential to address the pathology in patients. This is covered in more detail when an anaplerotic approach to correcting mitochondrial function is discussed later. While this defect may be downstream of the defect in ATM function, it may nevertheless be a useful target for the treatment of patients.
4. Metabolic Stress and A-T
A number of A-T features, including insulin resistance, are difficult to explain via a defect in the response to DNA damage. It seems likely that ATM-dependent stress pathways mediate susceptibility to metabolic syndrome, since chloroquine, which promotes ATM activity and inhibits the JNK stress kinase, increases sensitivity to insulin and decreases vascular disease [58]. Recently, an A-T patient cohort study demonstrated that diabetes is common, especially in older A-T patients, and often begins at puberty [59]. ATM deficiency is associated with insulin resistance and diabetes [60]. Specifically, an early study showed that the insulin-dependent dissociation of 4E-BP1 is significantly reduced in ATM-deficient cells [61]. The study went on to show that ATM phosphorylates 4E-BP1 at Ser 111 and that both in vitro and in vivo treatments with insulin induce this phosphorylation in an ATM-dependent manner, confirming the contribution of ATM to the metabolic abnormalities in A-T. Further, animal studies demonstrated that loss of one or both alleles of ATM increases the symptoms of metabolic syndrome, including glucose intolerance and insulin resistance in ApoE−/− mice fed on a high-fat diet [62]. They showed that during insulin resistance induced by ATM deficiency, JNK was activated, which consequently increased the level of inhibitory serine-307 phosphorylation on the insulin receptor substrate-1 (IRS-1) and caused insulin resistance. Other results suggest that lower ATM levels in a rat model fed a high-fat diet may contribute to the development of insulin resistance by downregulating Akt activity, since defective Akt activation is an important mechanism in the development of this resistance [60]. They also showed that in cells transfected with wild-type ATM, insulin caused a dramatic increase in the cell surface glucose transporter 4 (GLUT4), while in cells transfected with kinase-dead ATM, the translocation of GLUT4 to the cell surface in response to insulin was markedly inhibited. Therefore, the reduced PI3K/Akt signaling during ATM deficiency appears to contribute to the decrease in GLUT4 translocation and insulin resistance. However, it is evident that several pathways are involved in this signaling and the role of ATM is more complex. While these results offer a greater insight into metabolic syndrome, they raise the possibility that ATM has a greater role in regulating metabolism. A better understanding of this was obtained when it was shown that mice possessing a mutation in an ATM target site on p53 (S18A) showed increased metabolic stress, including increased inflammatory cytokines, reduced antioxidant gene expression and defects in glucose homeostasis, such as insulin resistance [62]. They also showed that deregulated ROS levels contributed to an imbalance in glucose homeostasis. As mentioned above, enhanced levels of ROS are a characteristic of A-T cells, suggesting that the well-established oxidative stress phenotypes of these cells, at least partially, contribute to metabolic stress [62].
It is evident that the efficient generation of ATP through glycolysis and mitochondrial oxidative phosphorylation is critical to cell survival. This is particularly acute for the survival of neurons, where the restoration of membrane potential after spiking is heavily ATP dependent [63]. Given the accumulating reports on mitochondrial dysfunction in a variety of A-T cell types, it might be predicted that this would have an impact on neurons in the CNS of A-T patients. Gene expression analysis in the cerebellar cortex predicted a generalized loss of nuclear-encoded mitochondrial proteins in A-T, emphasizing the importance of ATM in protecting mitochondrial function [63]. These data suggested that the demand for ATP might be more acute in the cerebellum of patients with A-T. They showed that the depletion of ATP generated ROS, which activated ATM and, in turn, led to the phosphorylation of nuclear respiratory factor 1 (NRF1) [64]. NRF1 was dimerized, translocated to the nucleus where it upregulated nuclear-encoded mitochondrial genes, enhancing the capacity of the electron transport genes and restoration of mitochondrial function. The replenishment of ATP was defective in Atm-deficient cells and chronic ATP depletion impacted on cell survival. The implication was that cerebellar cells would be exposed to an ATP deficit, which would contribute to their degeneration.
This entry is adapted from the peer-reviewed paper 10.3390/antiox11040653
This entry is offline, you can click here to edit this entry!