The first two marine BPs were isolated from the red algae Rhodomela larix. Bromophenol´s (BPs) have been isolated from red, brown and green algea, ascadians, mussels, marine proteobacteria and sponges. BPs are common marine secondary metabolites. BPs have been found to have many beneficial health properties.
The synthetic efforts have been concentrated on making more of isolated compounds, but also on improving the structures to obtain better biological effects. In that respect, it is of course useful to analyze the effects of already known compounds. It seems like the number of hydroxyl groups is an important factor and so is conjugation for anti-oxidant and anti-radical activity. Conjugation can be caused by nitro, acetyl or aldehyde groups preferentially in para-position to the OH-group. On the other hand, bromination does not always seem to be a determining factor.
- bromophenols
- synthesis
- biological effects
- biosynthesis
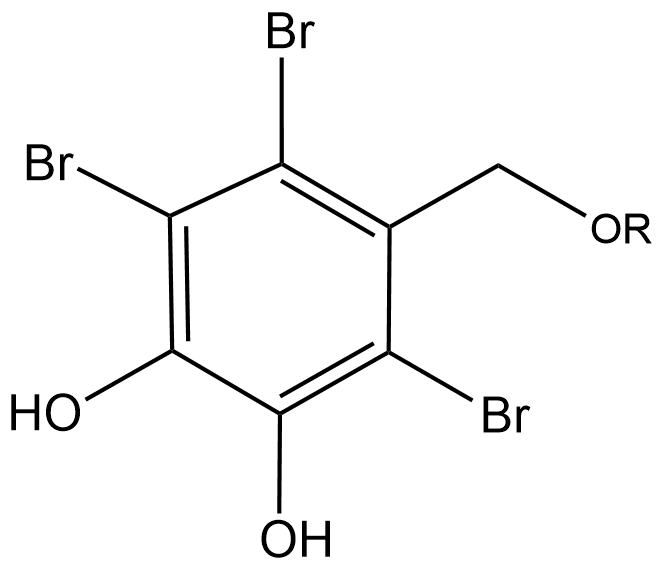
Figure 1. Typical bromophenol
The first two marine BPs were isolated from the red algae Rhodomela larix.[1] Bromophenol´s (BPs) have been isolated from red, brown and green algea, ascadians, mussels, marine proteobacteria and sponges. BPs are common marine secondary metabolites. BPs have been found to have many beneficial health properties.
The synthetic efforts have been concentrated on making more of isolated compounds, but also on improving the structures to obtain better biological effects. In that respect, it is of course useful to analyze the effects of already known compounds. It seems like the number of hydroxyl groups is an important factor and so is conjugation for anti-oxidant and anti-radical activity. Conjugation can be caused by nitro, acetyl or aldehyde groups preferentially in para-position to the OH-group. On the other hand, bromination does not always seem to be a determining factor. Other active conjugations are seen in biphenyl skeletons (Fig. 2) compared with no biphenyl type conjugation.
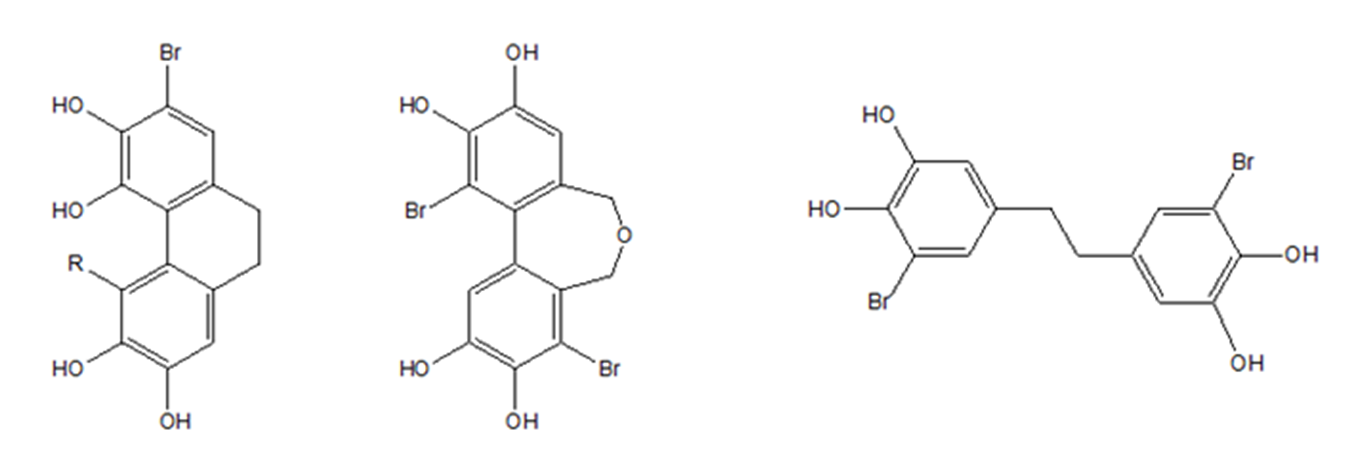
Figure 2. Biphenyl skeletons and a non-conjugated precursor (see later)
The synthesized compounds can roughly be categorized as six types as seen in Fig. 3.
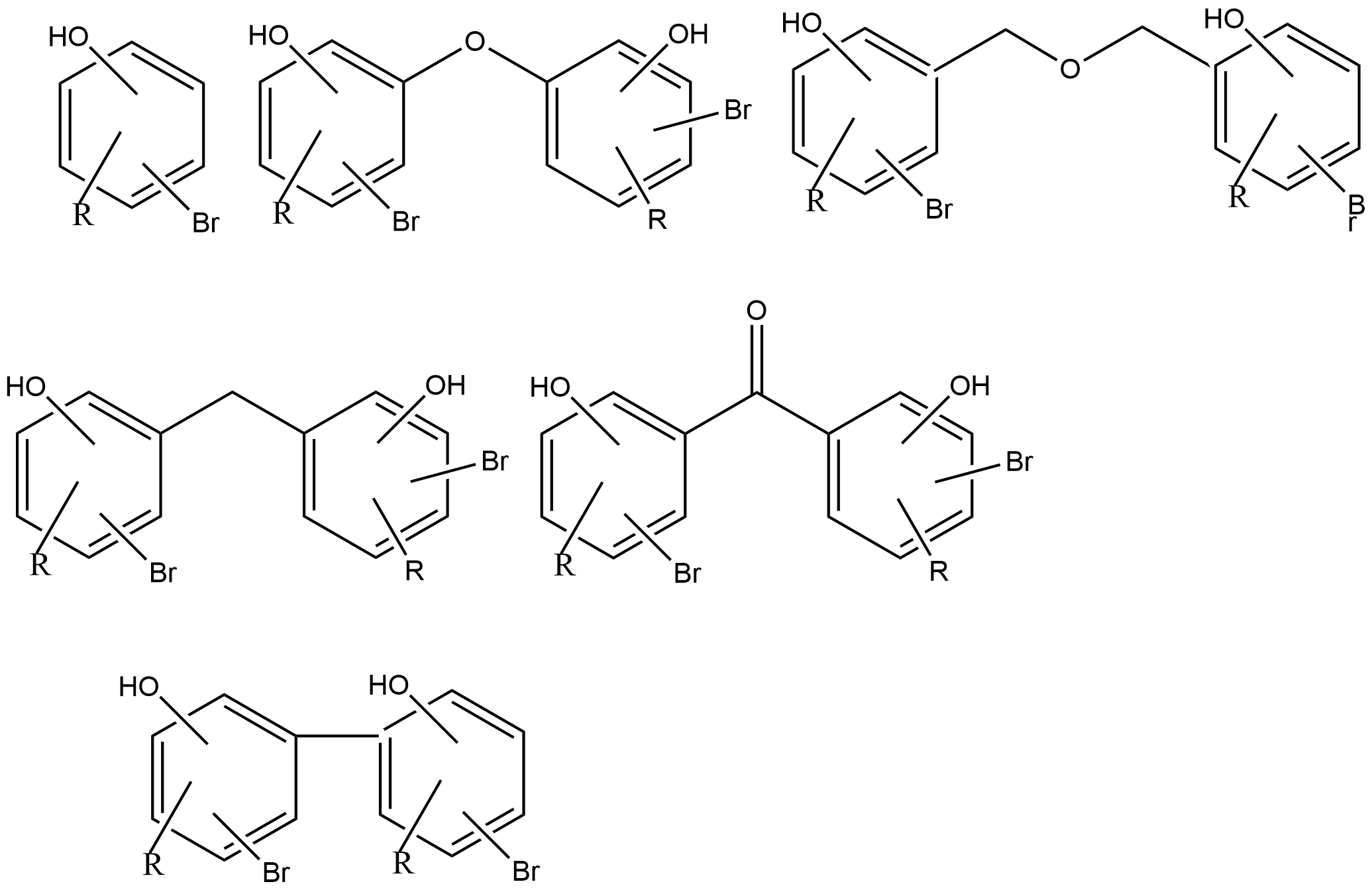
Figure 3. The six typical patterns. The number of OH, Br and substituents, R, may of course change and the pattern is not intended to indicate symmetrical compounds.
As will be seen in the following to achieve these natural product often many step synthesis is necessary, but normally using well known synthetic reactions. The OH groups often have to be protected. This is done by using OCH3 groups in the initial steps. These can then be removed using BBr3 or PBr3 (see later). Reviews, discussing bromophenols, are available and compounds isolated from the marine organisms mentioned above can be found in these two reviews. [2],[3]
1. Biosynthetic approaches
At present, polybrominated diphenyl ethers (PBDEs) are frequently used as brominated flame retardants, due to their durability and toxicity, PBDEs are regarded as persistent organic pollutants. The study showed the formation of hydroxylated polybrominated diphenyl ethers (HO-PBDEs) from simple bromophenols using bromoperoxidase. This provided a biosynthetic approach to synthesize HO-PBDEs. This biosynthetic approach using bromoperoxidase as a catalyst is seen in Fig .4. [4] This of course is also an indication that this could be a pathway in living organisms.
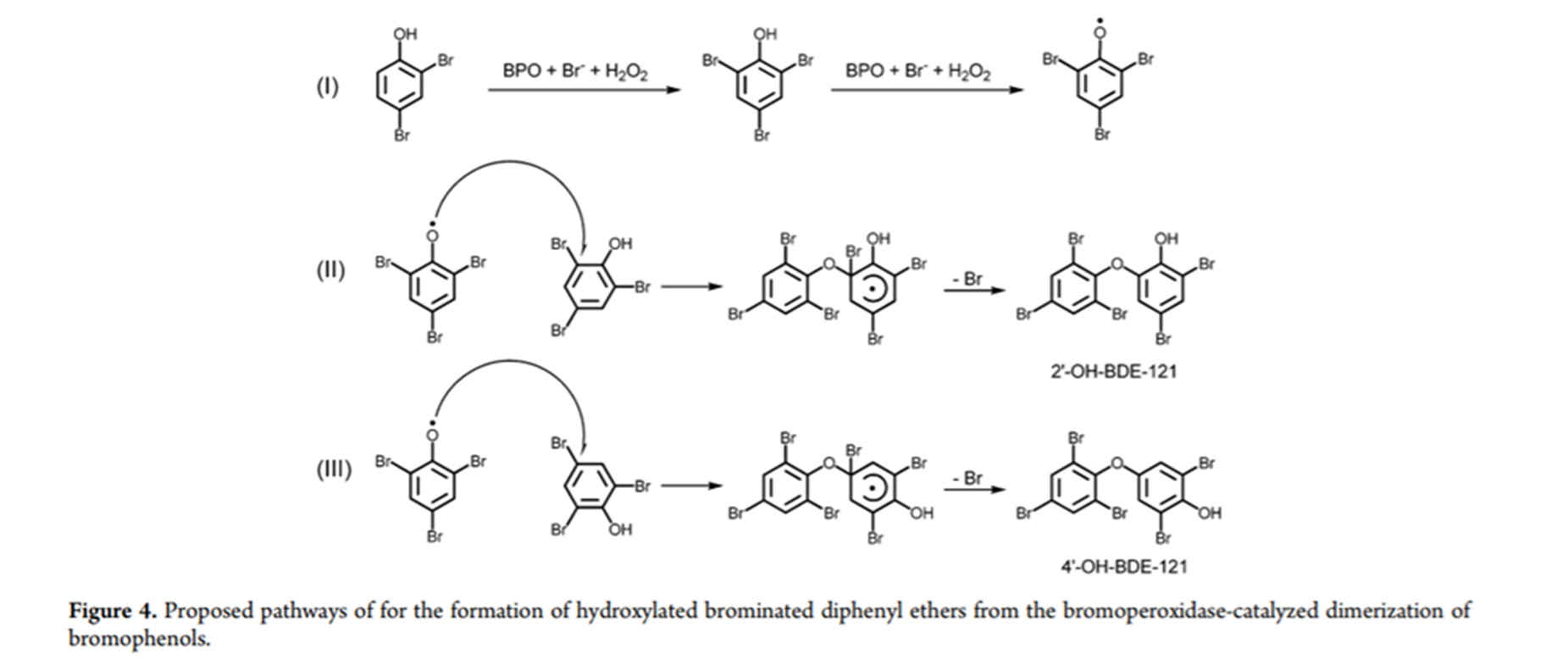
Figure 4. taken from Ref. 4 with permission from The American Chemical Society.
In another study laccase was demonstrated as the coupling catalyst.[5]
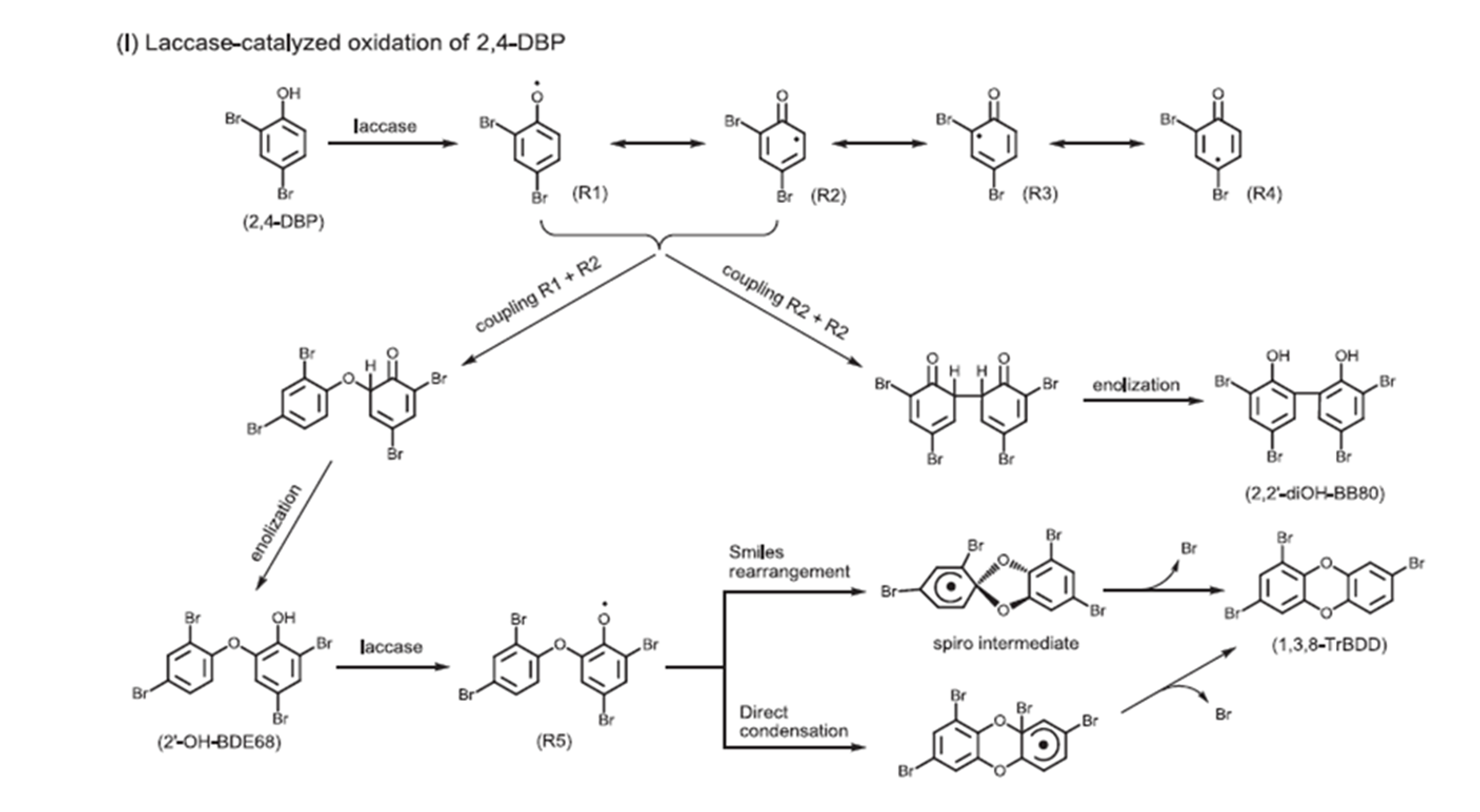
Figure 5. Use of laccase as coupling reagent. From Ref. 5 with permission from Elsevier.
2. Synthetic approaches
This study revealed the first synthesis of the natural products 1–3 and their derivatives with mono OMe 15–17 (Fig. 6). All these synthetic BPs exhibited effective inhibition of some metabolic enzymes, such as CA I and II, AChE, and BchE, illustrating that they can be used as good alternatives for the treatment of neurological disorders, glaucoma, epilepsy, mountain sickness, gastric and duodenal ulcers. Compounds 11-13 can be converted to the corresponding phenols (see Scheme 1 b of Fig. 6) [6]
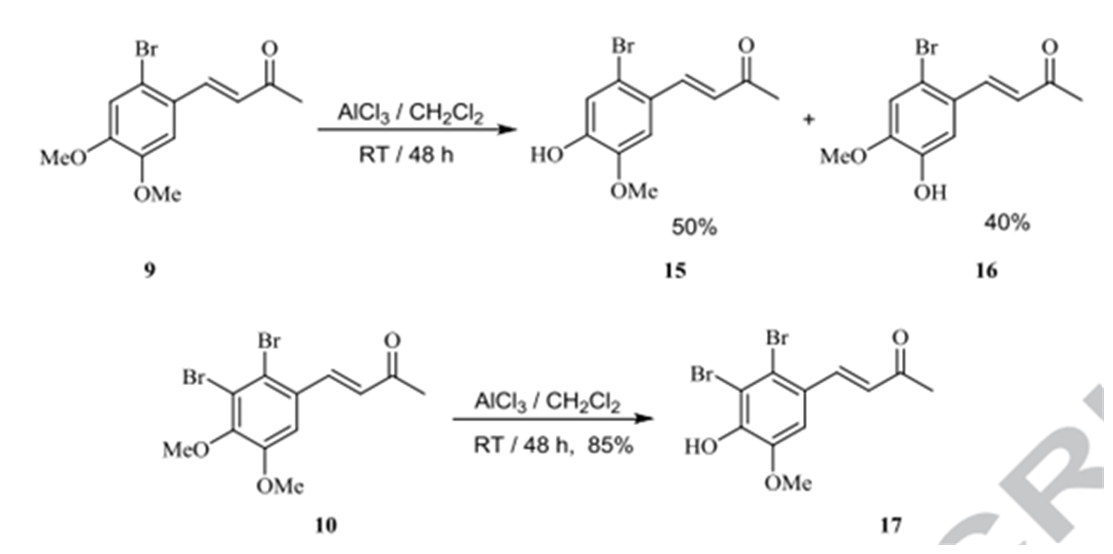
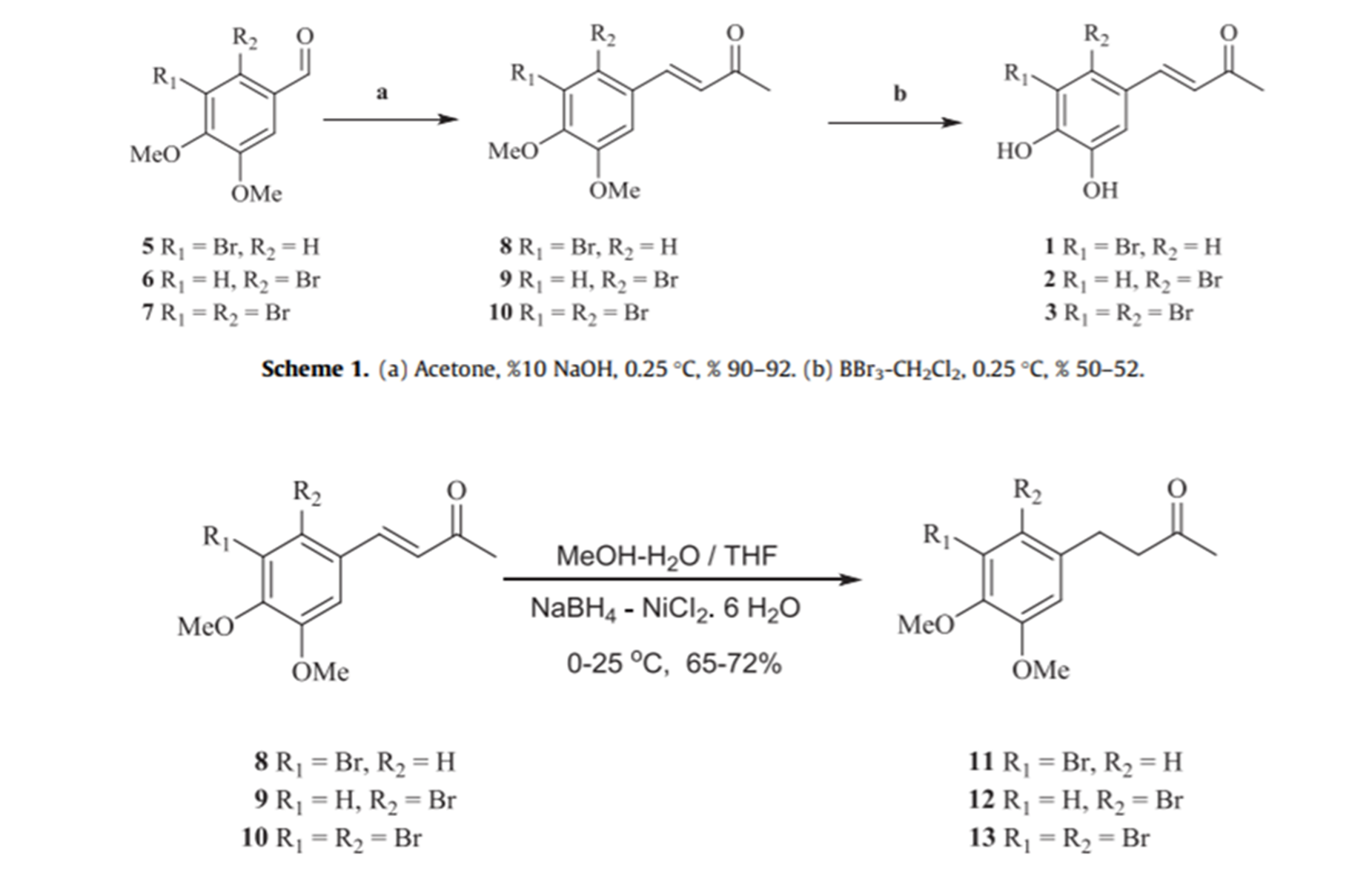
Figure 6. The lower part is Scheme 1b. Taken from Ref. 6 with permission from Elsevier.
In Ref. [7] a series of BP derivatives with CH3SO2 groups were synthesized and these compounds have anticholinergic activity. Among them, BPs 1 and 2 (Fig. 7a) inhibited the activity of AChE and BChE. Comparing these two compounds, BP1 with two hydroxyl groups and one bromine group had stronger activity towards BChE, whereas BP 2 with two hydroxyl groups and three bromine group had stronger activity towards AChE.
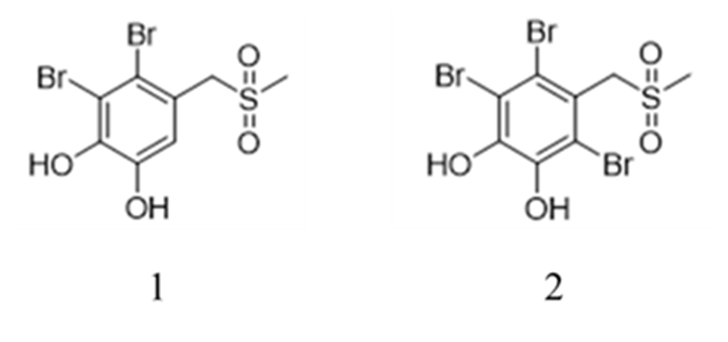
Figure 7a.
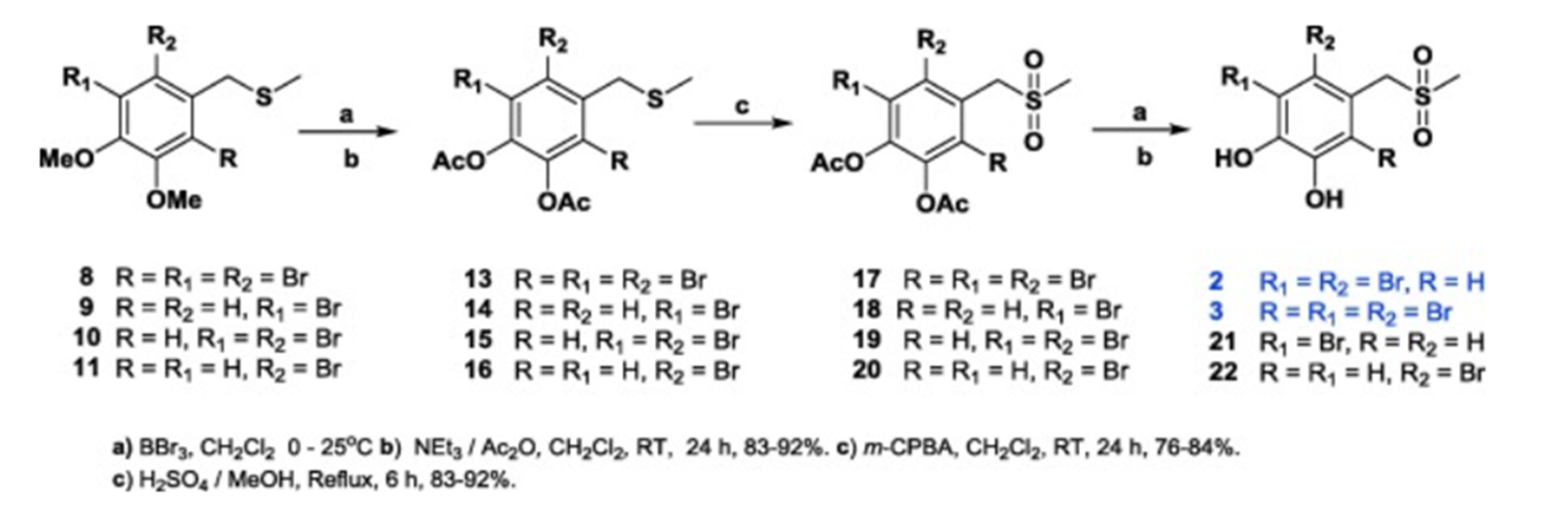
Figure 7b. Synthesis of sulphur containing BPs. Taken from Ref. 7 with permission from Elsevier.
Both enantiomers of Rhodomelin A (Fig. 8) were synthesized starting from larnosolaldehyde (4,5-dihydroxy-2,3-dintrobenzaldehyde). It incorporates a gamma-aminobutyric unit as well as a ureidopyrrolidone.[8]
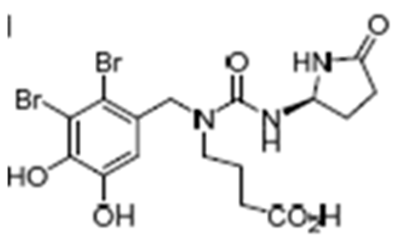
Figure 8. Rhodomelin A
Some synthetic derivatives containing indolin-2-one moiety have been designed and synthesized. [9] BPs 1-5 (see Fig. 9a) containing the indolin-2-one moiety displayed potent cytotoxicity to A549, Bel7402, HepG2, HeLa, and HCT-116 cancer cell lines, which is valuable for developing bromophenol derivatives as novel anticancer drugs.
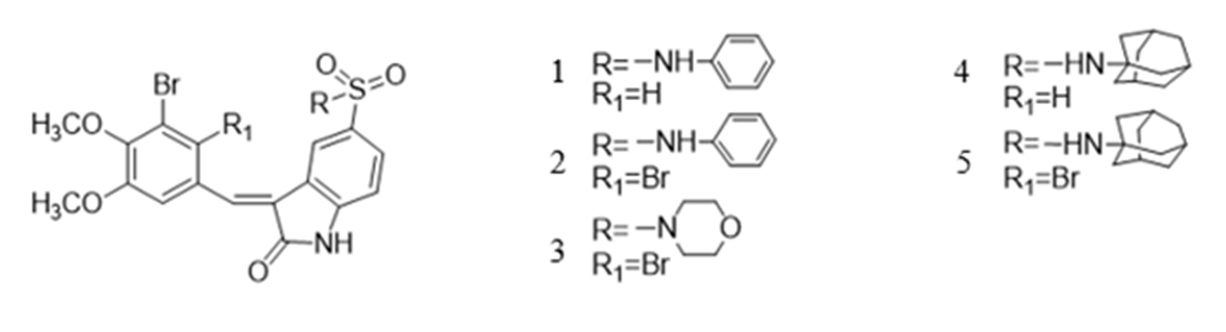
Figure 9a
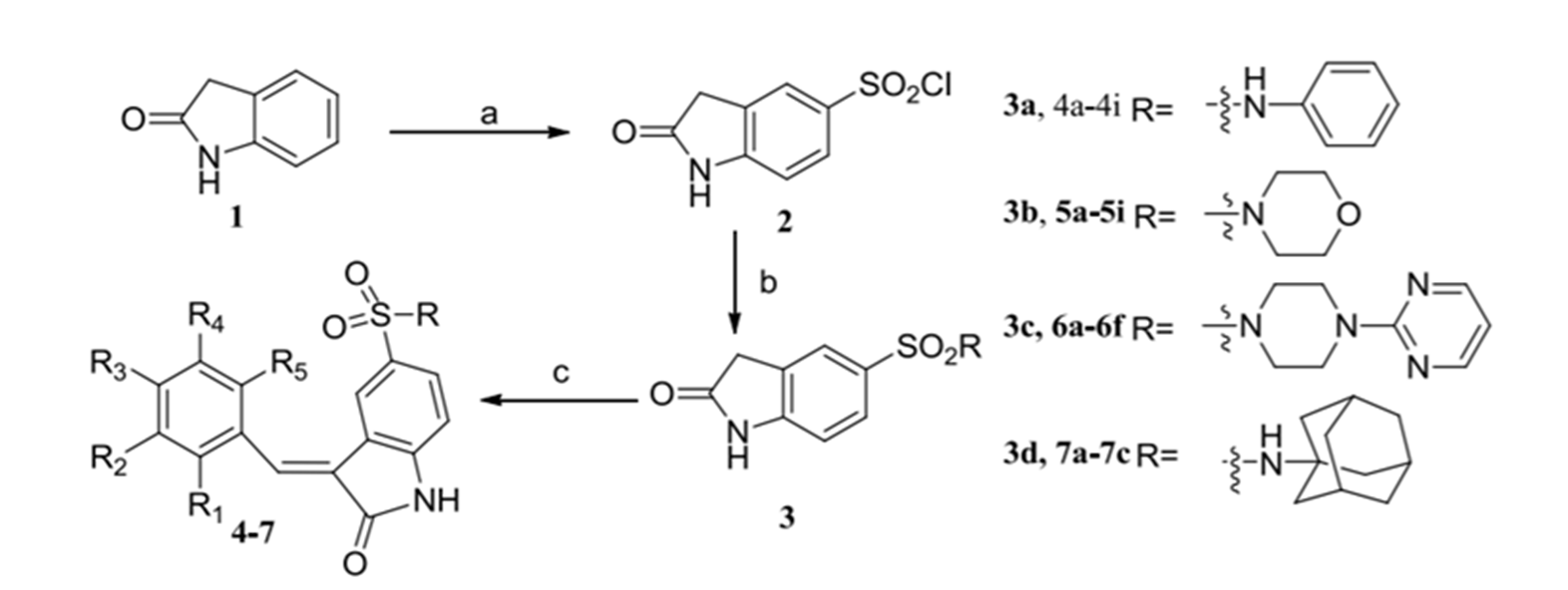
Figure 9. Synthesis of a BP with an indoline-2-one moity. Taken from Ref. 9.
The total synthesis of (±)-rhodoconferimide was carried out in this stydy. [10] Noticeably, (+)-rhodoconferimide showed more efficient antioxidant with IC50 of 5.22 μM compared with multifunctional urceolatin (IC50=7.90 μM) It may be a good antioxidant candidate for protection of humans from free-radical-induced damage.
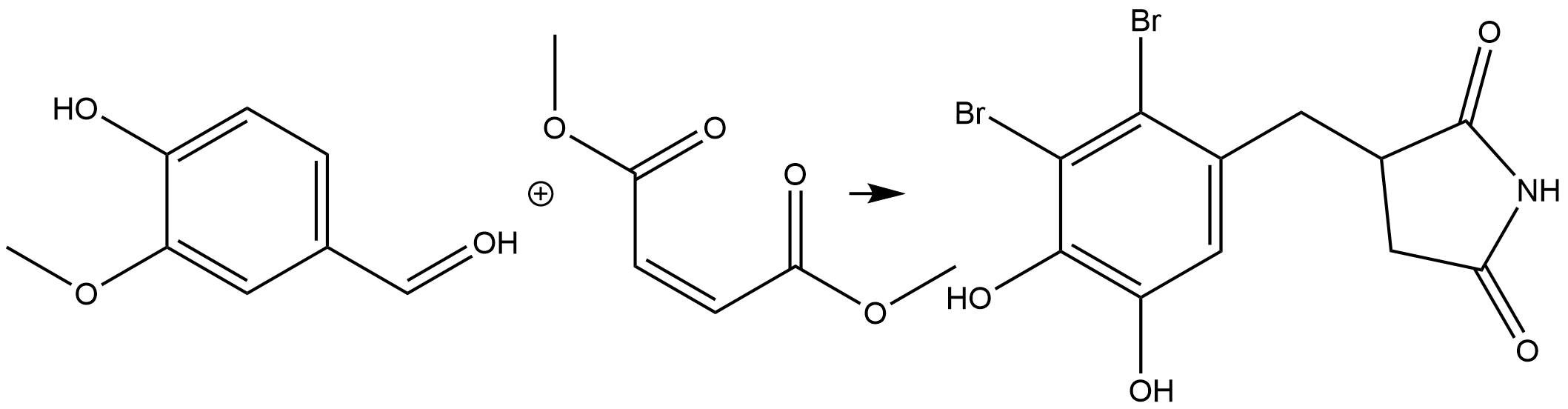
Figure 10 . Synthesis of Rhodoconferimide in seven steps.
A general way of synthesizing mono-bromophenols have been developed using electron-rich phenols, bromide ions, dioxygen and copper acetate.[11]
Bis(2,3-dibromo-4,5-dihydroxybenzyl) ether (BDDE) was synthesized and showed effective inhibition on α-glucosidase. When binding it induced minor conformational changes. This study established a useful pathway to obtain BDDE and confirmed its inhibition of α-glucosidase. It may be a potential agent for type 2 diabetes treatment.[12]
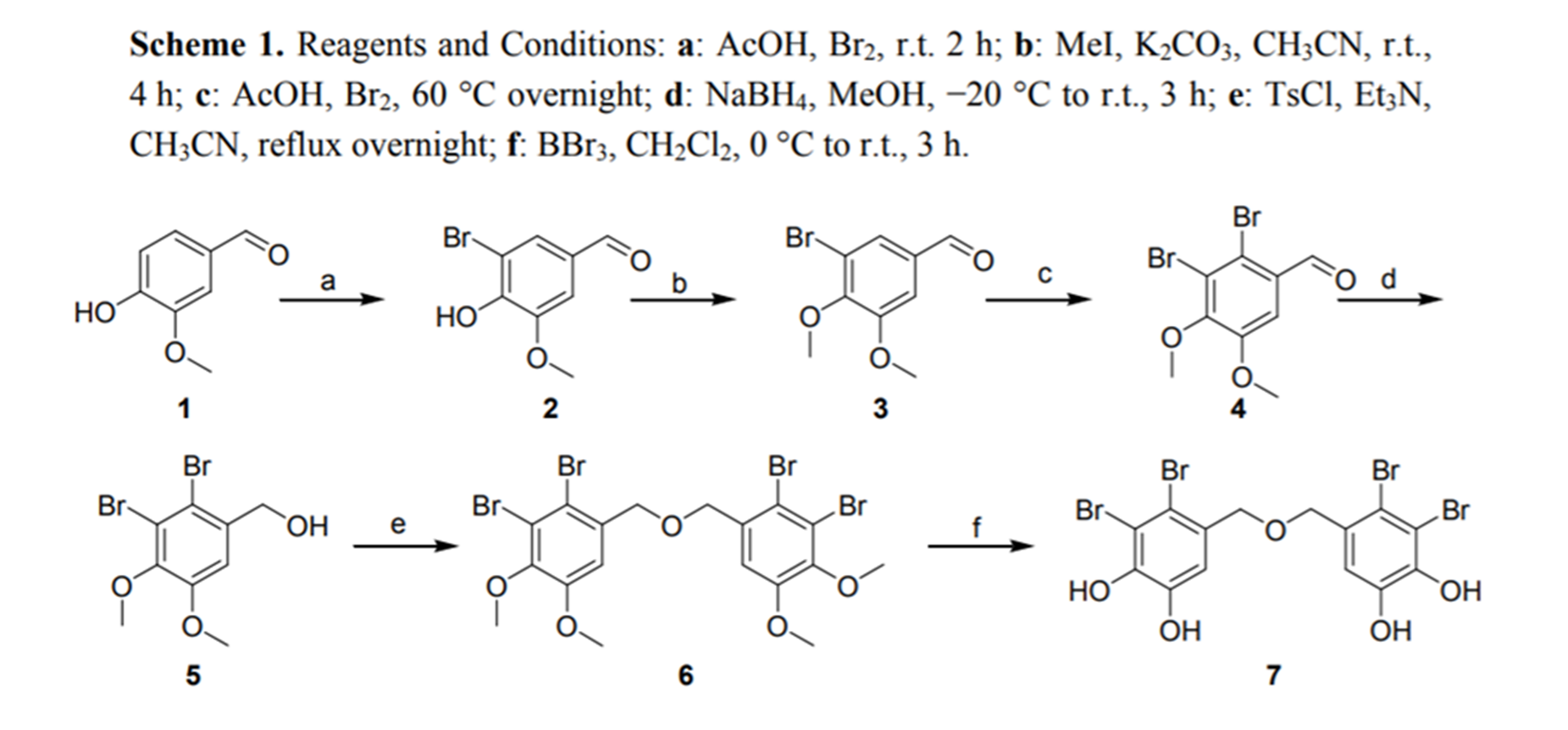
Figure 11 . Synthesis of an ether coupled BP. From Ref. 12.
Avrainvilleol was synthesized by coupling a tosylhydrazone of 2,3-methoxy-4-bromo-6-methoxymethylbenzaldehyde with the (3-brom o-4-mehoxyphenyl)boronic acid followed by demethylation leading to avrainvilleol, 1 (Fig. 12).[13]
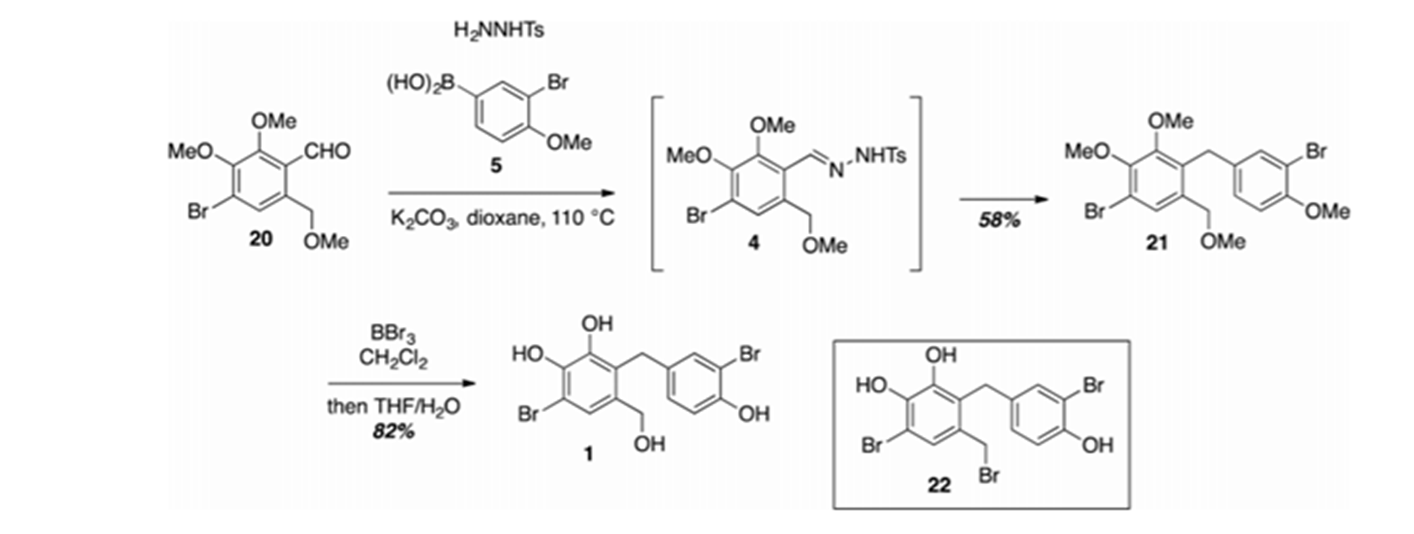
Figure 12. Synthesis of avrainvilleol. From Ref. 13 with permission from The America Chemical Society.
A different attempt to create a PhCH2Ph unit was achieved by Guo et al. using a Friedel-Crafts reaction [14] (Fig. 13). The protein tyrosine phosphatase 1B (PTP1B) inhibitory activities of the synthetic compounds were evaluated by the colorimetric assay. The results showed that these compounds are moderate PTP1B inhibitors.
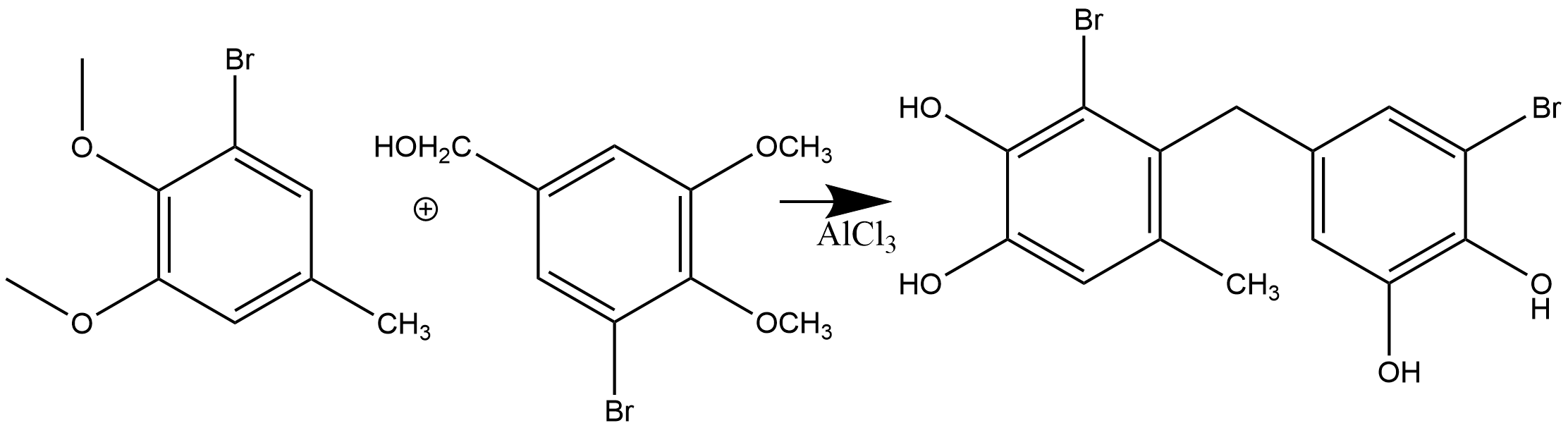
Figure 13. The total synthesis is in eight steps.
In Ref. [15], a series of compounds that inhibit hCA have been synthesized. Among them, BP 22 (Fig. 14) is a natural product isolated from the Caribbean red algae Vidalia obtusaloba, and had strong inhibitory effect on hCA I, II, IV, and VI. Whereas BP 21, which was the fully methylated derivative of BP 22, had weaker inhibition against hCA I, II, IV, and VI, indicating the importance of the -OH groups in the activity against hCA.
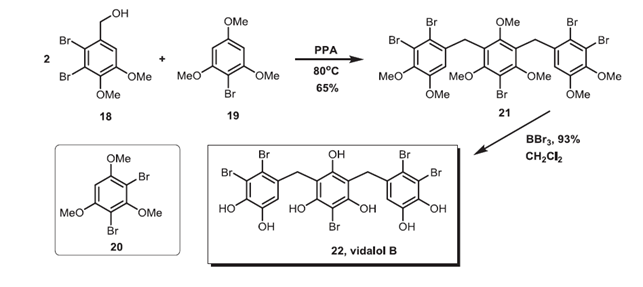
Figure 14. Synthesis of vidalol B. From Ref. 15 with permission from Elsevier.
In Ref. [16] based on the natural BP 1, four O-methylated derivatives BPs 2-5 (Fig. 15) were synthesized and showed powerful hCA I, II, IV, and VI inhibitory activity. The structures of BPs 2 and 4 are cis, whereas those of BPs 3 and 5 are trans. The comparison of these four compounds indicated that the difference in configuration had little effect on the hCA inhibitory activity.
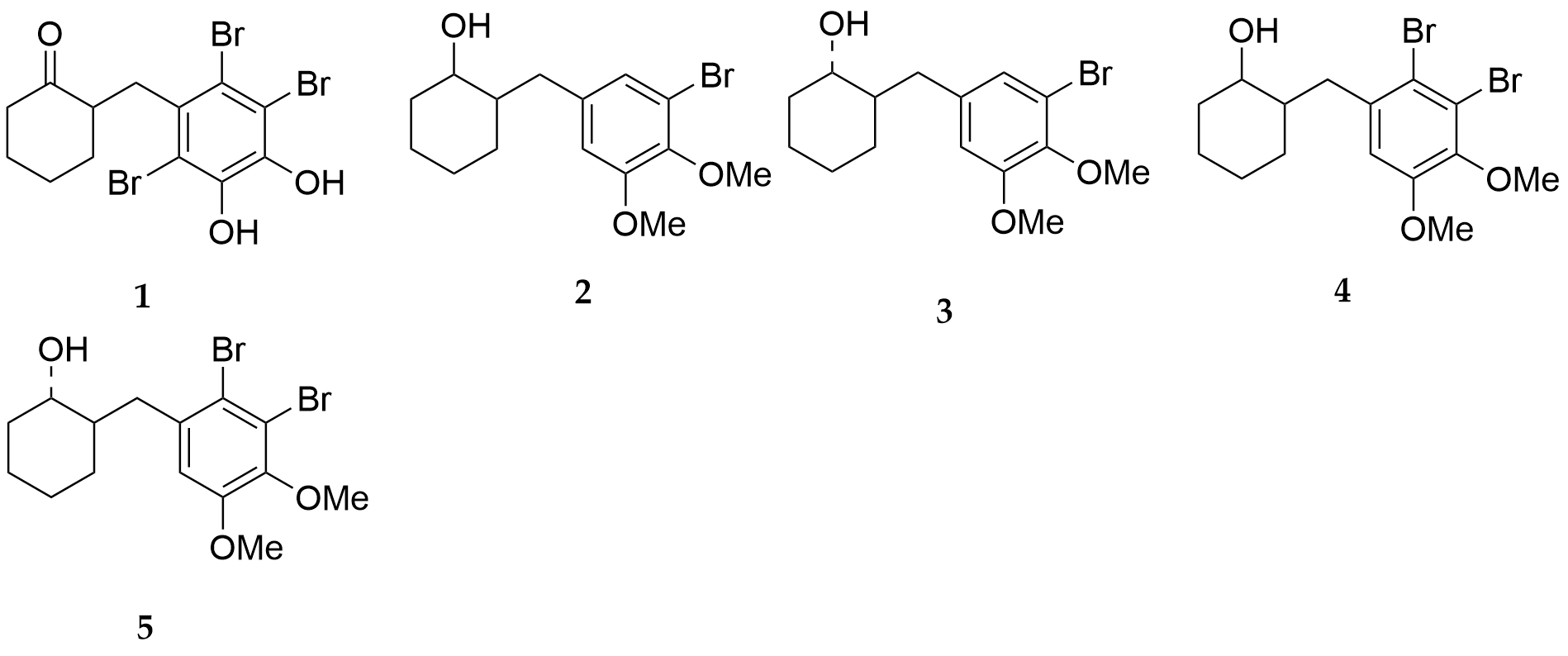
Figure 15. Cyclohexanol derivatives
In Ref. [17] the natural product BP 1 was synthesized (Fig. 16), revealing inhibitory activity for hCA I and II. Based on this, the same research group also synthesized four BPs 2-5 which exhibited enhanced inhibitory activity for hCA I and II. Compared with BPs 3-5, BP 2 showed better inhibition activity towards hCA I and II, suggesting an attenuator effect of the bromine moiety. When compared with the natural compound BP 1 with hydrophobic groups, BPs 2-5 containing hydroxyl groups had stronger inhibition activity of hCA I and II.
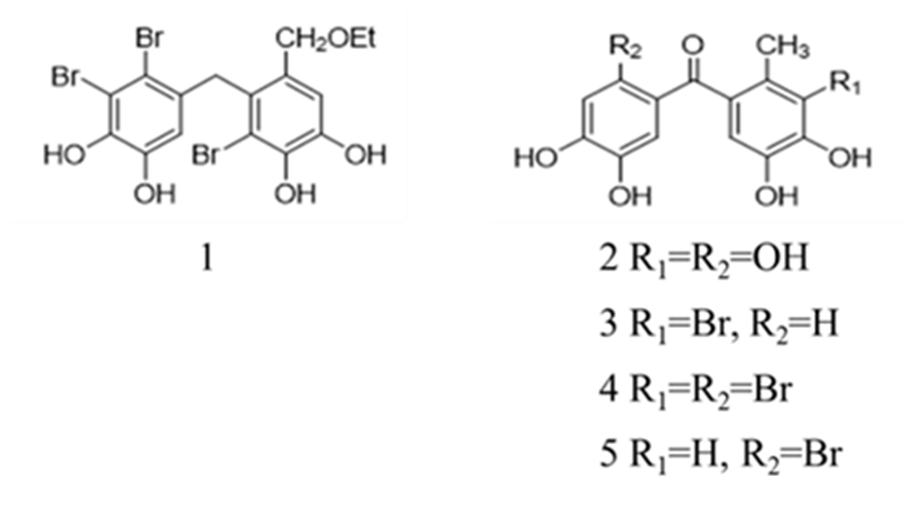
Figure 16.
In Ref. [18] based on some natural compounds, new compounds, which could inhibit hCA II, have been synthesized. Among them, BPs 1 and 2 (Fig. 17), obtained from red algae Symphyocladia latiuscula, had weak inhibitory effects on human carbonic anhydrase (hCA) II However, the new derivative 3 (Figure 17) had a stronger activity (IC50 = 0.7 µM). This indicates that the carbonyl group is very important for inhibiting the activity of hCA II.
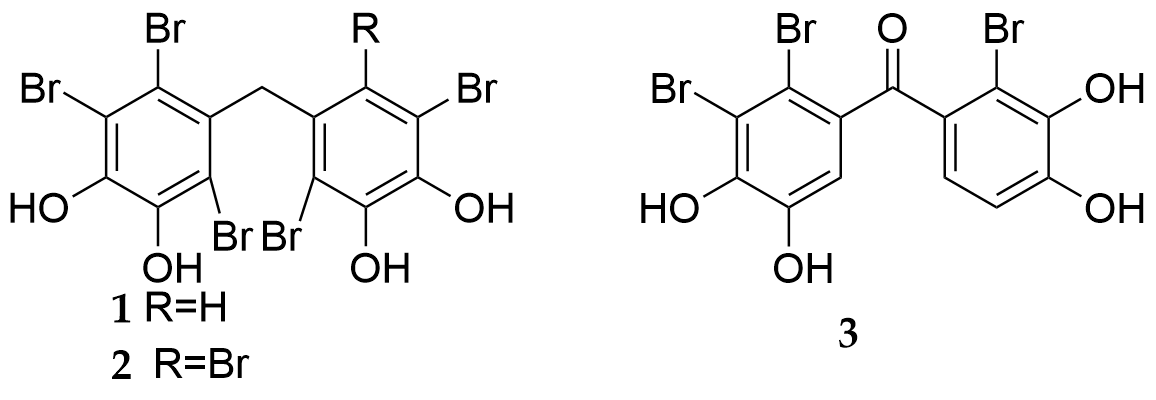
Figure 17.
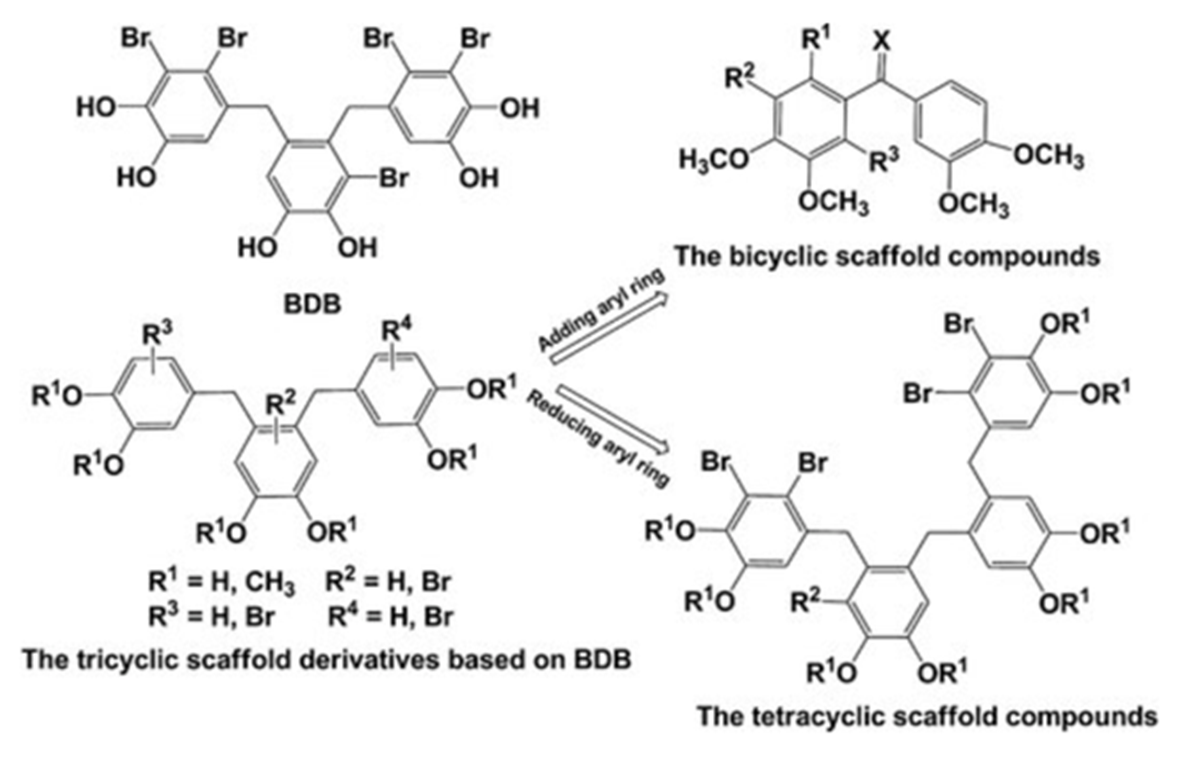
Figure 18 . Synthesis of a tetracyclic scaffold. From Ref. [19] with permission from Wiley.
In Ref. 19 a new compound, which could inhibit PTP1B activity was synthesized as described in Fig. 18. BP 2 (Fig. 19) was a synthetic derivative of the natural BP 1. BP 2 also inhibited PTP1B (IC50 = 0.89 µM), and its activity was about two-fold higher compared with the lead compound 1 (IC50 = 1.7 µM). A preliminary SAR study revealed that the tricyclic scaffold and multi-bromine atoms (four to five) attached to the aryl rings were critical for PTP1B inhibition.
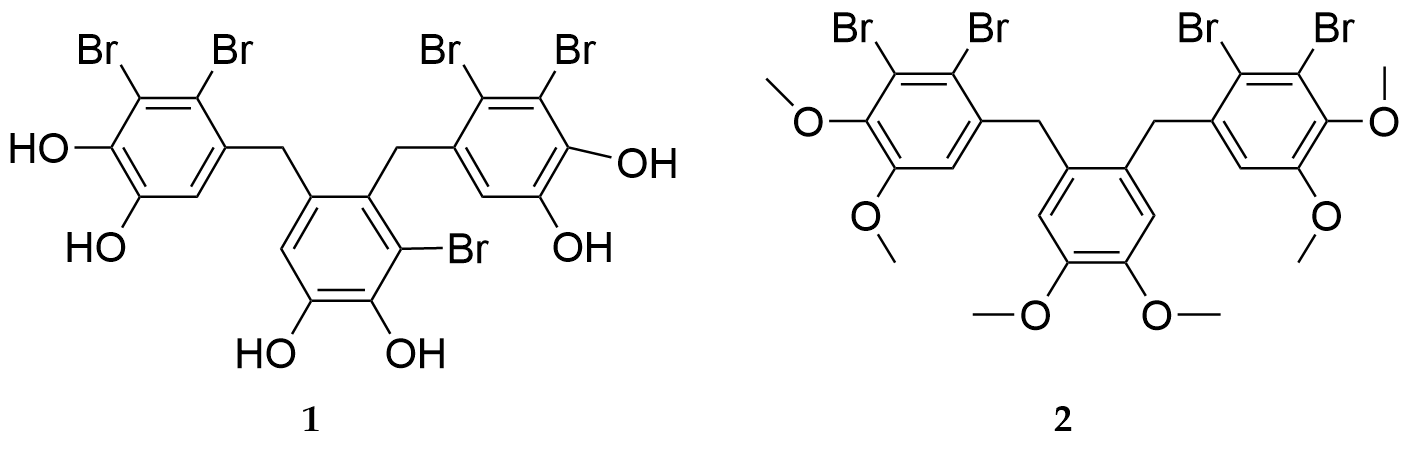
Figure 19.
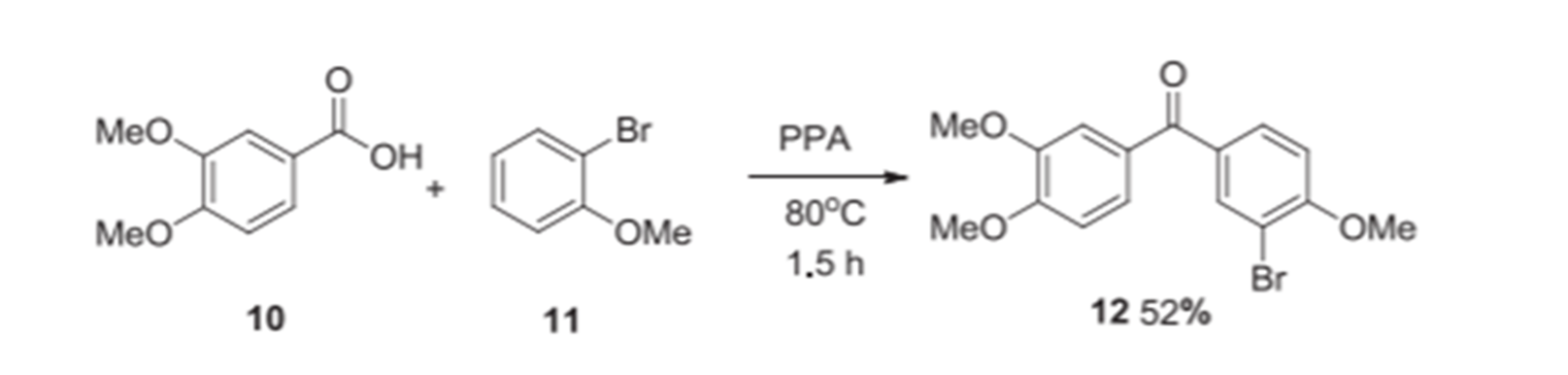
Figure 20. Synthesis of benzophenone type BPs. Taken from Ref. [20] with permission from Elsevier.
In Ref. 20 the new BP derivatives 1-7 (Fig. 21), which have acetylcholinesterase and α-glycosidase enzymes inhibition properties and antioxidant activity been synthesized among them, BP 3 exhibited the strongest inhibitory effect on α-glycosidase, indicating these have antidiabetic activity. These compounds could effectively clear DPPH free radicals, but have weaker ability to scavenge ABTS radical. They also showed AChE inhibitory activity, among them, BP 5 which have two bromines in para-position and one methoxy, showed the strongest inhibitory ability of AChE.
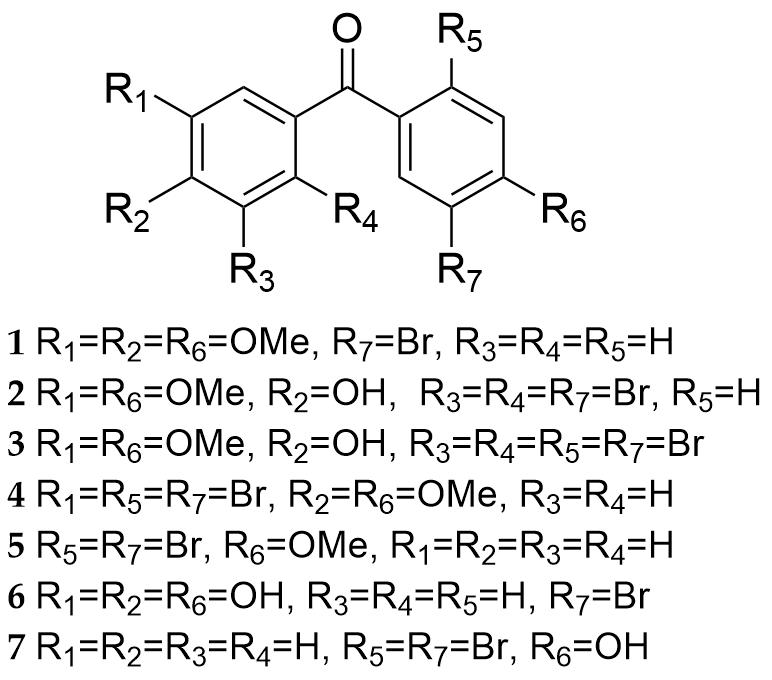
Figure 21.
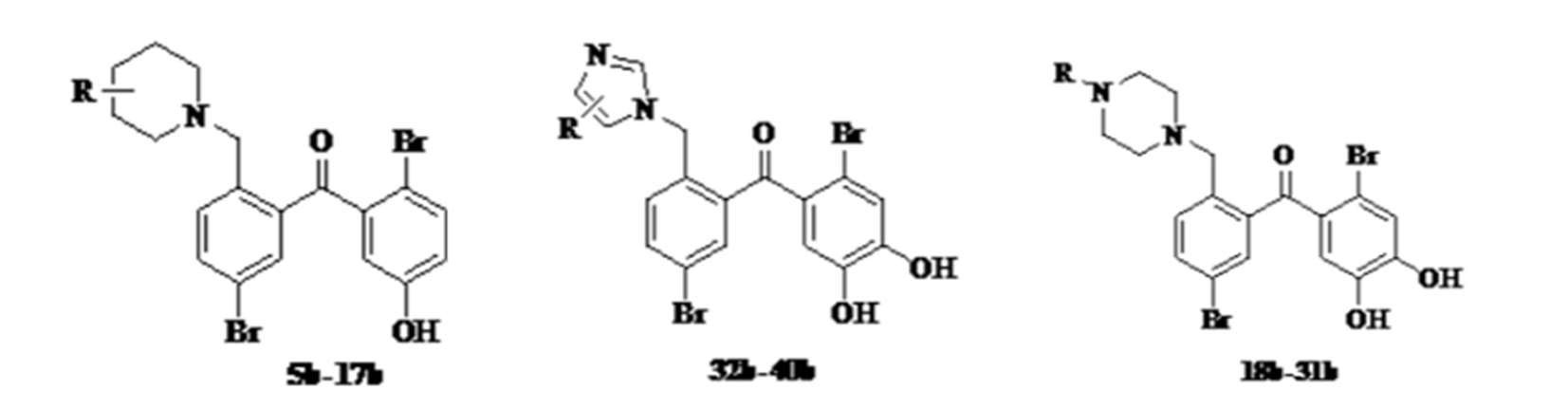
Figure 22. Synthesis of piperidine, imidazole and pyrazine derivatives.
The synthesis was achieved in several steps starting from 2-methyl-5-bromobenzoic acid, which is converted into the acid choride to form the coupling in a Friedel-Crafts reaction. [21]
When the series of nitrogen-containing heterocycles such as piperidine, piperazine, and imidazole replaced the OH group at the 2-position of 1 (Fig. 23), the products also showed moderate-to-potent cytoprotective activity against H2O2-induced injury in EAhy926 cells with 2 being the most potent. A SAR study revealed that the antioxidant ability of these analogues increased with an increasing number of heterocycles and hydroxyl groups. Furthermore, a molecular-docking study demonstrated that compound 2 could interact with Keap1, and thus in turn modulating Keap1-Nrf2 protein-protein interaction in order to activate Nrf2-induced downstream protective genes from oxidative stress damage.
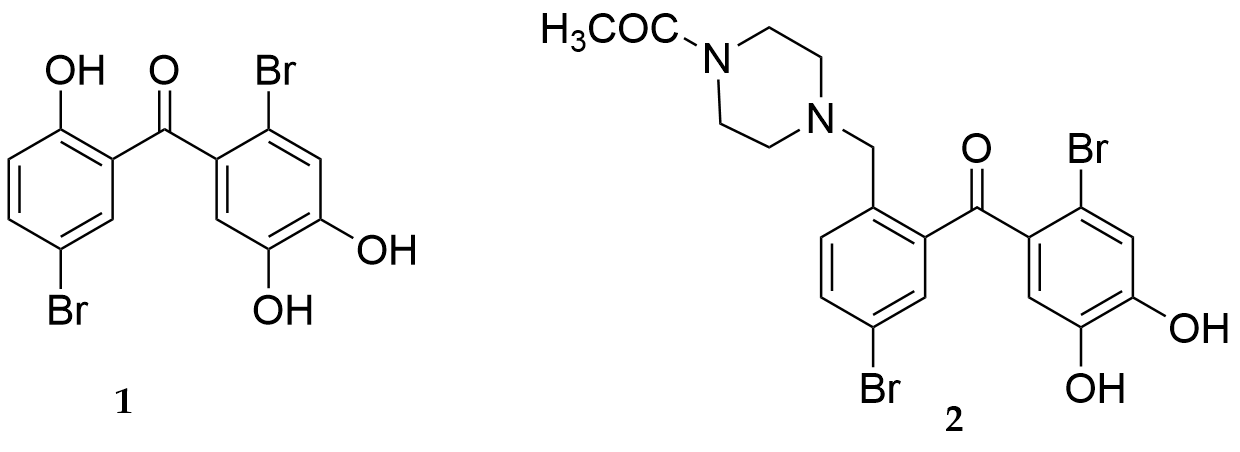
Figure 23.
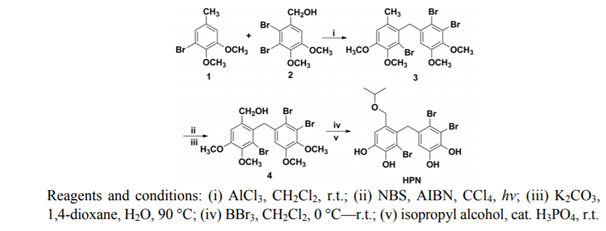
Figure 24. From Ref. [22]
One of the new HPN compounds (Fig. 25) showed in in vitro experiments, that this compound could inhibit the activity of PTP1B and had high selectivity against other PTPs. In in vivo test, the compound could effectively reduce the serum triglyceride and total cholesterol concentration in the serum of mice after 8 weeks of action, and the results of an intraperitoneal glucose tolerance test in Sprague–Dawley rats indicate, that this compound had a similar antihyperglycemic activity as rosiglitazone. Furthermore, this compound could decrease PTP1B levels in pancreatic tissue, indicting HPN had the potential to treat type 2 diabetes. Ref. 22.
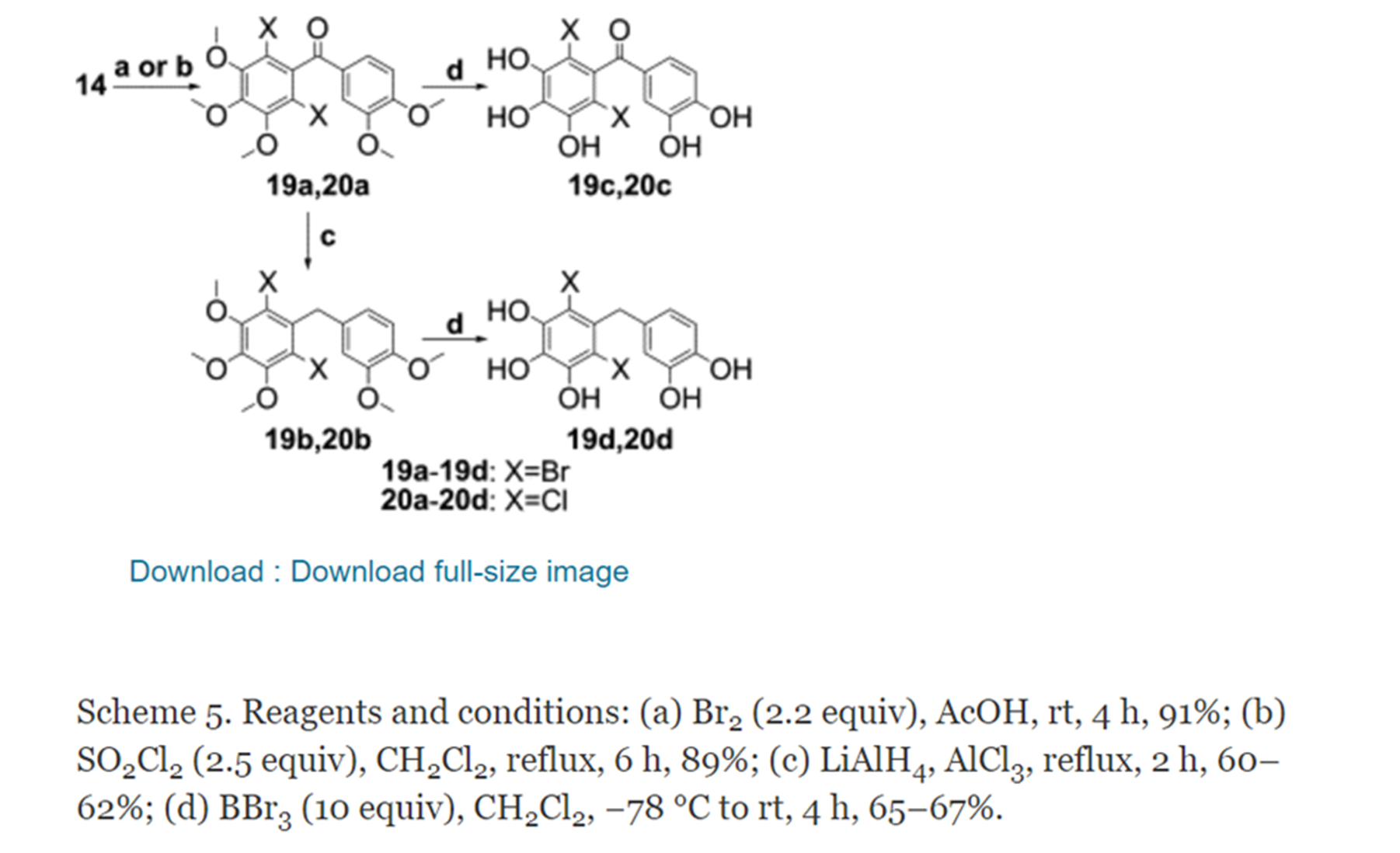
Figure 25. From Ref. with permission from Elsevier.
In Ref. [23] a number of new halophenols were synthesized (Fig. 26). Among them, BP 1and 2 (Fig. 27) showed excellent antioxidant activity. They were also able to protect human umbilical vein endothelial cells from H2O2-induced oxidative stress injury. Through the comparison of a series of synthesized compounds, it is found that hydroxyl groups on the diphenyl backbone are essential for the in vitro antioxidative activities of these compounds, and the presence of one or more halogen atoms on the phenol ring is also necessary. The number and position of the hydroxyl groups and halogen atoms may influence the potency of activity.
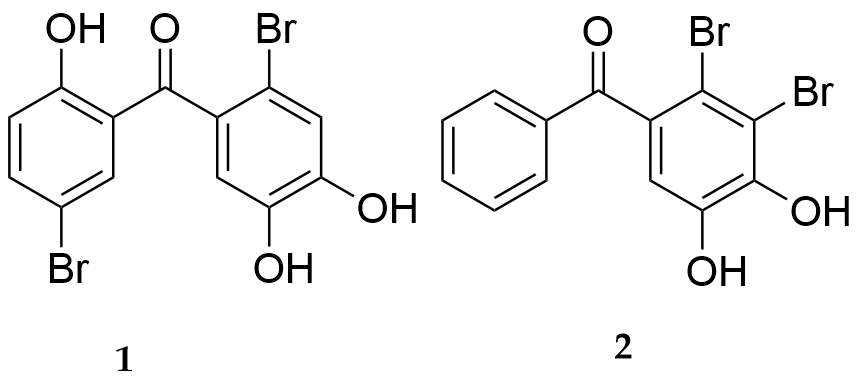
Figure 26.
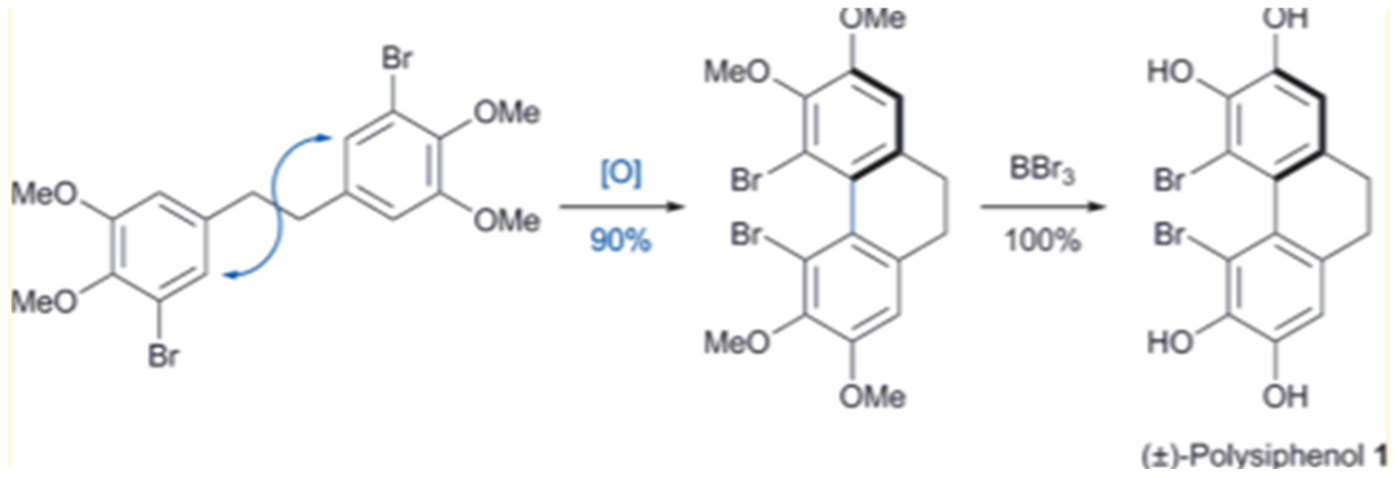
Figure 27. Synthesis of polysiphenol. Taken from Ref. [24] with permission from The American Chemical Society.
+- Polysiphenol was obtained by oxidative coupling followed by demethylation.
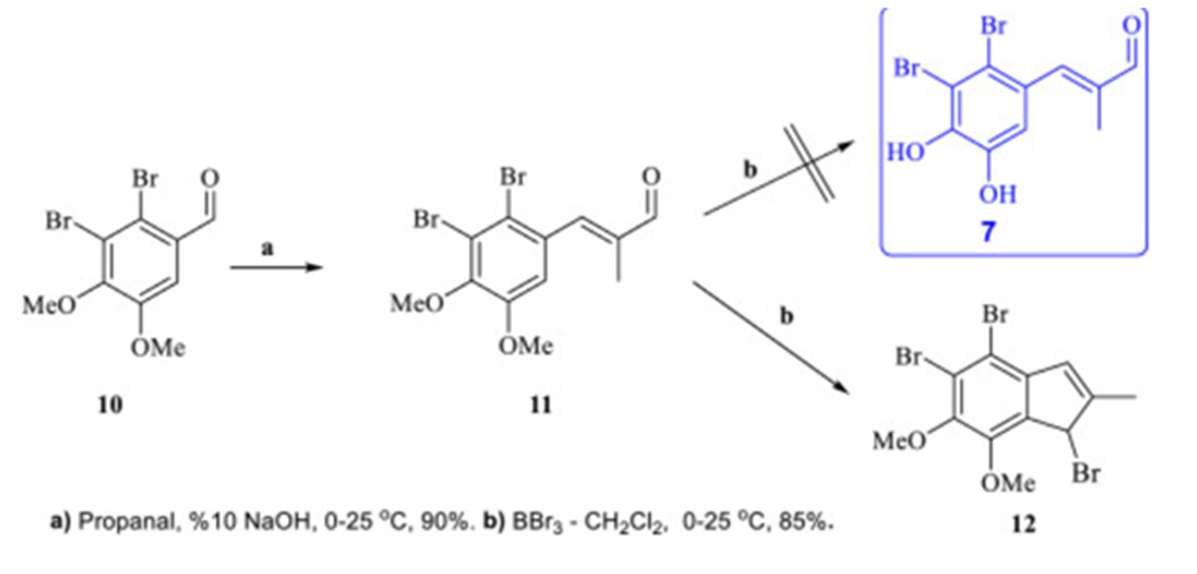
Figure 28. Synthesis of an indene derivative. Taken from Ref. [25] with permission from Elsevier.
This entry is adapted from the peer-reviewed paper 10.3390/md18080411
References
- Wang, S.; Wang, L.J.; Jiang, B.; Wu, N.; Li, X.; Liu, S.; Luo, J.; Shi, D.; Anti-angiogenic properties of BDDPM, a bromophenol from marine red alga Rhodomela confervoides, with multi receptor tyrosine kinase inhibition effects.. Int. J. Mol. Sci. 2015, 16, 13548-13560, .
- Liu, M.; Hansen, P.E.; Lin, X.; Bromophenols in marine algae and their bioactivities.. Mar. Drugs. 2011, 9, 1273-1292, .
- Hui Dong; Songtao Dong; Poul Erik Hansen; Dimitrios Stagos; Xiukun Lin; Ming Liu; Progress of Bromophenols in Marine Algae from 2011 to 2020: Structure, Bioactivities, and Applications. Marine Drugs 2020, 18, 411, 10.3390/md18080411.
- Lin,K.; Gan,J. and Liu,W.; Production of Hydroxylated Polybrominated Diphenyl Ethers from Bromophenols by Bromoperoxidase-Catalyzed Dimerization. . Environ. Sci. Technol. 2014, 48, 11977−11983, .
- Kunde Lin; Shiyang Zhou; Xi Chen; Jiafeng Ding; Xiaoyan Kong; Jay Gan; Formation of hydroxylated polybrominated diphenyl ethers from laccase-catalyzed oxidation of bromophenols. Chemosphere 2015, 138, 806-813, 10.1016/j.chemosphere.2015.08.014.
- Çetin Bayrak; Parham Taslimi; Ilhami Gulçin; Abdullah Menzek; The first synthesis of 4-phenylbutenone derivative bromophenols including natural products and their inhibition profiles for carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase enzymes. Bioorganic Chemistry 2017, 72, 359-366, 10.1016/j.bioorg.2017.03.001.
- Çetin Bayrak; Parham Taslimi; Halide S. Karaman; Ilhami Gülçin; Abdullah Menzek; The first synthesis, carbonic anhydrase inhibition and anticholinergic activities of some bromophenol derivatives with S including natural products. Bioorganic Chemistry 2019, 85, 128-139, 10.1016/j.bioorg.2018.12.012.
- Li, K.; Wang, Y.F.; Li, X.M.; Wang, W.J.; Xu, T; Isolation, synthesis, and radical-scavenging activity of rhodomelin A, a ureidobromophenol from the marine red alga Rhodomela confervoides.. Org. Letters. 2018, 20, 417-420, .
- Li-Jun Wang; Shuaiyu Wang; Bo Jiang; Ning Wu; Xiangqian Li; Bao-Cheng Wang; Jiao Luo; Meng Yang; Shui-Hua Jin; Da-Yong Shi; et al. Design, Synthesis and Biological Evaluation of Novel Bromophenol Derivatives Incorporating Indolin-2-One Moiety as Potential Anticancer Agents. Marine Drugs 2015, 13, 806-823, 10.3390/md13020806.
- Pandhade, K.R. and Argade, N.P.; First Total Synthesis of (±)-Rhodoconferimide. Synthesis 2018, 50, 658-662, .
- L. Menini; L. A. Parreira; E. V. Gusevskaya; A Practical Highly Selective Oxybromination of Phenols with Dioxygen.. ChemInform 2007, 38, 6401-6404, 10.1002/chin.200751085.
- Ming Liu; Wei Zhang; Jianteng Wei; Xiukun Lin; Synthesis and α-Glucosidase Inhibitory Mechanisms of Bis(2,3-dibromo-4,5-dihydroxybenzyl) Ether, a Potential Marine Bromophenol α-Glucosidase Inhibitor. Marine Drugs 2011, 9, 1554-1565, 10.3390/md9091554.
- Aaron R. Wegener; Kenneth A. Miller; Total Synthesis of Avrainvilleol. The Journal of Organic Chemistry 2017, 82, 11655-11658, 10.1021/acs.joc.7b02028.
- Guo, S; Li, J.Li, T Shi, D and Han, L.; Synthesis of three bromophenols form red algae as PTP1B inhibitors. Chin.J.Ocean.Limnol. 2011, 29, 68-74, .
- Halis T. Balaydın; Murat Şentürk; Süleyman Göksu; Abdullah Menzek; Synthesis and carbonic anhydrase inhibitory properties of novel bromophenols and their derivatives including natural products: Vidalol B. European Journal of Medicinal Chemistry 2012, 54, 423-428, 10.1016/j.ejmech.2012.05.025.
- Halis T. Balaydın; Murat Şentürk; Abdullah Menzek; Synthesis and carbonic anhydrase inhibitory properties of novel cyclohexanonyl bromophenol derivatives. Bioorganic & Medicinal Chemistry Letters 2012, 22, 1352-1357, 10.1016/j.bmcl.2011.12.069.
- Yusuf Akbaba; Halis T. Balaydın; Abdullah Menzek; Süleyman Göksu; Ertan Sahin; Deniz Ekinci; Synthesis and Biological Evaluation of Novel Bromophenol Derivatives as Carbonic Anhydrase Inhibitors. Archiv der Pharmazie 2013, 346, 447-454, 10.1002/ardp.201300054.
- Halis Türker Balaydın; Hakan Söyüt; Deniz Ekinci; Süleyman Göksu; Sukru Beydemir; Abdullah Menzek; Ertan Sahin; Synthesis and carbonic anhydrase inhibitory properties of novel bromophenols including natural products. Journal of Enzyme Inhibition and Medicinal Chemistry 2011, 27, 43-50, 10.3109/14756366.2011.574131.
- Bo Jiang; Da-Yong Shi; Yongchao Cui; Shuju Guo; Design, Synthesis, and Biological Evaluation of Bromophenol Derivatives as Protein Tyrosine Phosphatase 1B Inhibitors. Archiv der Pharmazie 2012, 345, 444-453, 10.1002/ardp.201100373.
- Necla Öztaşkın; Rüya Kaya; Ahmet Maraş; Ertan Şahin; Ilhami Gülcin; Süleyman Göksu; Synthesis and characterization of novel bromophenols: Determination of their anticholinergic, antidiabetic and antioxidant activities. Bioorganic Chemistry 2019, 87, 91-102, 10.1016/j.bioorg.2019.03.010.
- Xiu E. Feng; Qin Jin Wang; Jie Gao; Shu-Rong Ban; Qing-Shan Li; Synthesis of Novel Nitrogen-Containing Heterocycle Bromophenols and Their Interaction with Keap1 Protein by Molecular Docking. Molecules 2017, 22, 2142, 10.3390/molecules22122142.
- Da-Yong Shi; Shuju Guo; Bo Jiang; Chao Guo; Tao Wang; Luyong Zhang; Jing-Ya Li; HPN, a Synthetic Analogue of Bromophenol from Red Alga Rhodomela confervoides: Synthesis and Anti-Diabetic Effects in C57BL/KsJ-db/db Mice. Marine Drugs 2013, 11, 350-362, 10.3390/md11020350.
- Wanyi Zhao; Xiue Feng; Shurong Ban; Wenhan Lin; Qing‐Shan Li; Synthesis and biological activity of halophenols as potent antioxidant and cytoprotective agents. Bioorganic & Medicinal Chemistry Letters 2010, 20, 4132-4134, 10.1016/j.bmcl.2010.05.068.
- Tim N. Barrett, D. Christopher Braddock, Anna Monta, Michael R. Webb, and Andrew J. P. White J.; Total Synthesis of the Marine Metabolite -Polysiphenol via Highly Regioselective Intramolecular Oxidative Coupling. Nat. Prod. 2011, 74, 1980–1984, .
- Cetin Bayrak; Abdullah Menzek; The first synthesis of phenylpropanoid derivative bromophenols including natural products: Formation of an indene derivative compound. Tetrahedron 2020, 76, 131016, 10.1016/j.tet.2020.131016.