Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Integrative & Complementary Medicine
There is strong evidence from geographical ecological and observational studies that solar UVB exposure and serum 25-hydroxyvitamin D concentration are inversely correlated with risk of incidence and death from many types of cancer. Clinical trials have provided limited support for the UVB-vitamin D-cancer hypothesis due to poor design and execution. Many experimental studies in cultured cells and animal models have described a wide range of anticancer effects of vitamin D compounds.
- vitamin D
- 25-hydroxyvitamin D
- 1,25-(OH)2D3
- antitumor action
- breast cancer
- case-control studies
- colorectal cancer
1. Introduction
The epidemiology of vitamin D and cancer can be examined through the prisms of ecological studies, observational studies, and clinical trials. This review looks at findings from ecological studies of cancer risk with respect to indices of solar ultraviolet-B (UVB) doses, observational studies of cancer risk with respect to serum 25(OH)D concentration and oral vitamin D intake, and randomized controlled trials (RCTs) of cancer risk with respect to vitamin D supplementation.
Epidemiological data prompted the study of the putative anticancer action of vitamin D in the laboratory. Two important considerations in the study of the action of 1,25-(OH)2D3 and analogues in experimental cancer systems are the expression of vitamin D receptor (VDR), which is frequently low or absent, and the high doses of its ligands that are usually required to observe effects. A lack of VDR is linked to transcriptional (by silencing by DNA methylation or repression by SNAIL1/2), posttranscriptional (by several microRNAs) or posttranslational (phosphorylation, alteration of subcellular localization) inhibitory mechanisms, and low cell responsiveness to VDR ligands is often associated with upregulation of the 1,25-(OH)2D3 degrading enzyme CYP24A1 in tumor cells.
2. Perspectives on Epidemiological Studies
2.1. Ecological Studies
As would be generally expected, incidence and mortality rates are generally inversely correlated with solar UVB indices unless UVB exposure is linked to increased risk, such as that for melanoma and other skin cancer. The direct correlation with oral cavities and the pharynx in the United States is consistent with UVB exposure’s being a risk factor for lip cancer. Solar UVB exposure increases human papillomavirus (HPV) concentrations, as evidenced by peak rates of positive Pap smears for cervical cancer in Denmark in August [103]. HPV is a risk factor for head and neck cancer [104]. HPV is also hypothesized to be an important risk factor for melanoma [105].
The finding that the incidence rates for several cancers are directly correlated with solar UV in China, whereas most of the cancer mortality rates are inversely correlated, is probably owing to the fact that air pollution levels are much higher in northern than in southern China [106]. In addition, vitamin D generally reduces the risk of cancer mortality rates rather than incidence rates. The reasons may include that although many factors affect cancer incidence, few factors affect cancer progression and metastasis.
Because the countries included are different in many respects, including diet, ethnicity, latitude, and pollution level, ecological studies offer strong evidence that UVB irradiance affects cancers similarly regardless of many other factors.
An important reason why ecological studies have shown robust relationships between indices of solar UVB doses is that they included many cases of cancer. Researchers conducting earlier ecological studies were more likely than researchers of more recent studies to find significant correlations with UVB doses because people back then spent more time in the sun without concern for skin cancer or photoaging, and obesity rates were lower.
2.2. Observational Studies
Several findings are important from the analyses presented regarding observational studies.
First, the inverse relationships between serum 25(OH)D concentration and cancer incidence or mortality rates are similar to those between solar UVB and cancer reported in ecological studies. The primary exception is for head and neck cancer; serum risk was inversely correlated with both serum 25(OH)D concentration and vitamin D intake. However, ecological studies showed direct correlations between solar UVB and both incidence and mortality rates for oral cavity/pharynx and pharynx cancers, although one study reported an inverse relationship for laryngeal cancer [25].
Secondly, a long follow-up time was again found to significantly decrease the observed beneficial effect of 25(OH)D concentration. For example, the meta-analysis of CRC risk with respect to 25(OH)D concentration by Hernandez-Alonso and colleagues [72] had 11 studies (one CC, nine NCC, and one meta-analysis) and six prospective cohort studies. The OR for the CC study was 0.45 (95% CI, 0.36–0.57). For the nine NCC studies, the mean follow-up time was near 8 years, and the OR was 0.63, whereas for the prospective cohort studies, the mean follow-up time was 13 years, and the OR was 0.80 (95% CI, 0.66–0.97).
Some parties have argued that CC studies with 25(OH)D concentration measured near the time of diagnosis would be the best type of observational study due to possible reverse causality [53]. There is no evidence to indicate that having undiagnosed cancer reduces 25(OH)D concentration other than perhaps decreasing with the progression cancer stage. Thus, CC studies, which are easier to conduct than prospective studies, are preferred.
The epidemiological and mechanical evidence regarding solar UVB exposure and vitamin D presented here generally satisfy Hill’s criteria for causality in a biological system (based on Kosh’s postulates) [107,108,109]. The only weakness is that RCTs have not yielded strong support, largely because they were poorly designed and conducted. However, as argued by Dr. Thomas R. Frieden, former head of the U.S. Centers for Disease Control and Prevention, in a review in The New England Journal of Medicine, RCTs have substantial limitations [110]. The review tabulates the strength and limitations of 11 study designs, including RCTs, prospective cohort, retrospective cohort, case-control, and ecological studies. It concludes by stating that there is no single, best approach to the study of health interventions, and clinical and public health decisions are almost always made with imperfect data.
2.3. Historical Overview
Many of the articles reviewed regarding epidemiological studies of solar UVB dose or exposure and vitamin D played important roles in developing the understanding of the role of vitamin D in reducing risk of cancer incidence and mortality rates. Table 10 lists a few of them in chronological order. Note that the importance of some of the articles, notably those reported prior to 1980, was not recognized until many years later.
Table 10. List of epidemiological studies that had important findings in the history of solar UVB exposure and/or vitamin D and cancer.
| Year | Finding | Reference |
|---|---|---|
| 1936 | Sun exposure can cause skin cancer but reduce risk of internal cancer. | [1] |
| 1937 | US Navy personnel highly exposed to sun had high skin cancer rates but low internal cancer rates. | [2] |
| 1941 | Cancer mortality rates for whites in the U.S. found inversely related to a solar radiation index while skin cancer (melanoma) mortality rates were directly related. | [3] |
| 1980 | Annual solar radiation dose inversely correlated with colon cancer mortality rate, USA, vitamin D production suggested. | [6] |
| 1985 | Dietary vitamin D and calcium inversely correlated with colorectal cancer incidence. | [7] |
| 1989 | Serum 25(OH)D concentration inversely correlated with colon cancer incidence. | [8] |
| 1990 | Annual solar radiation dose inversely correlated with breast cancer mortality rate in the U.S. | [9] |
| 2002 | Mortality rates for thirteen types of cancer are inversely correlated with solar UVB doses in the U.S., 1970–1994. | [13] |
| 2006 | A Harvard cohort study finding that incidence of several types of cancer were inversely correlated with predicted 25(OH)D concentration. | [111] |
| 2006 | An ecological study in the U.S. finding that incidence and mortality rates for many types of cancer were inversely correlated with solar UVB doses. | [22] |
| 2007 | A meta-analysis presenting a 25(OH)D concentration-colorectal cancer incidence relationship. | [46] |
| 2007 | An RCT conducted in the U.S. finding that vitamin D supplementation significantly reduced risk of all-cancer incidence rate. | [95] |
3. Stromal Effects: Cancer-Associated Fibroblasts
Today, the critical role of stroma in the carcinogenic process is clear. Fibroblasts are the main cellular component of tumor stroma (Cancer-Associated Fibroblasts, CAF). This is a heterogeneous cell population of multiple origins (tissue-resident fibroblasts, myeloid precursors, pericytes and adipocytes, among others) and features that is acquired via the change to an “activation phenotype”. It is thought to promote cancer invasion, angiogenesis and metastasis; inhibit the immune response; and reduce intratumoral delivery and the activity of chemotherapeutic agents [244,245]. However, the protective effects of CAF have also been described in some systems, and reprogramming their phenotype is accepted as a more advisable strategy than their elimination [246,247]. Early studies showed that VDR agonists have antifibrotic and antitumoral effects by antagonizing TGF-β in the intestine, liver, and pancreas [248,249,250,251,252].
1,25-(OH)2D3 regulated over one hundred genes in human CAF isolated from tumor biopsies of five breast cancer patients [253]. The induced gene signature reflects an antiproliferative and anti-inflammatory effect of 1,25(OH)2D3. Importantly, 1,25-(OH)2D3 inhibits the protumoral action of human colon CAF by reprograming them to a less activated phenotype. Thus, 1,25(OH)2D3 reduces the capacity of CAF to alter the ECM and their ability to promote the migration of colon carcinoma cells [254]. 1,25-(OH)2D3 regulates over one thousand genes in colon CAF that are involved in cell adhesion, differentiation and migration, tissue remodeling, blood vessel development, and the inflammatory response. Remarkably, 1,25(OH)2D3 imposes a gene signature that correlates with a better prognosis for colon cancer patients [254]. Curiously, in contrast to the antagonism reported in colon carcinoma cells, 1,25-(OH)2D3 and Wnt3A have an additive, partially overlapping effect in colon fibroblasts [255,256]. In line with the results in colon CAF, 1,25(OH)2D3 decreases the amount of miR-10a-5p found in the exosomes secreted by human pancreatic CAF, which attenuates the promigratory and pro-invasive effects that these CAF exert on pancreatic carcinoma cells [257]. Of note, a recent study reported that calcipotriol promotes an antitumorigenic phenotype of pancreatic CAF by reducing the release of prostaglandin (PG) E2, IL-6, periostin, and other factors. However, it reduces T-cell-mediated immunity [258]. Clearly, the action of VDR agonists on fibroblasts associated with distinct human cancers is a highly interesting, open line of research.
4. Effects on Cancer Stem Cells
Cancer stem cells (CSC) are supposedly a small population of cells present in tumors that are responsible for tumor initiation, growth, malignization, metastasis, and resistance to therapies. They originate from the mutational and epigenetic alteration of normal stem cells that maintain the homeostasis of tissues in adult life and behave as a source of new functional differentiated cells following injuries or in aging. The characterization and study of CSC present two unresolved problems: (a) the lack of confirmed universal or even tissue-specific markers, and (b) the existence of cell plasticity in tumors that implies differentiation/dedifferentiation processes during tumorigenesis and thus the lack of a stable stem phenotype but, instead, interconversion of stem and non-stem cells.
At present, there are two systems to study CSC: organoid cultures generated by CSC present in patient-derived tumor biopsies and subcultures of established, immortal tumor cell lines enriched in populations of cells expressing putative CSC markers and/or selected by their capacity to grow in suspension. Clearly, fresh, primary organoids are a more valuable system. They are three-dimensional (3D), self-organized multicellular structures generated by normal stem cells or CSC (that allow matched normal and tumor organoids to be obtained from a patient) that grow embedded in an ECM covered by a complex, tissue-specific, usually serum-free medium [259,260]. Organoids recapitulate some of the features of a particular organ or tumor of origin and are quite stable genetically, and thus are considered a better system to study cancer processes than 2D cell lines grown for decades on plastic dishes [261]. 1,25(OH)2D3 profoundly and differentially regulates the gene expression profile of colon cancer patient-derived normal and tumor organoid cultures. 1,25(OH)2D3 induced stemness-related genes (LGR5, SMOC2, LRIG1, and others) in normal but not tumor organoids [262]. In both normal and tumor organoids, 1,25(OH)2D3 reduced cell proliferation and the expression of proliferation and tumorigenesis genes that affected only a few Wnt/β-catenin target genes (MYC, DKK4). Importantly, 1,25(OH)2D3 induced some features of epithelial differentiation in tumor organoids cultured in proliferation medium, such as microvilli, adhesion structures, partial chromatin condensation, and increased cytoplasmic organelles. These effects were also observed in rectal tumor organoids [263].
Concordantly, 1,25(OH)2D3-regulated genes were involved in cell proliferation, differentiation, adhesion, and migration in another study using patient-derived colon organoids [264]. Moreover, MDL-811, an allosteric activator of the sirtuin (SIRT)6 deacetylase, reduced cell proliferation in colon carcinoma cell lines and patient-derived organoids and has a synergistic antitumoral effect in combination with vitamin D in Apcmin/+ mice [265]. However, conflicting data have been found in normal, nontumoral organoids: whereas 1,25-(OH)2D3 increased the stemness genes and the undifferentiated associated cell phenotype in organoids from healthy colon and rectum tissues of a dozen individuals [262,263], it enhanced the differentiation of organoids established from a benign region of a radical prostatectomy from a single patient [266].
A series of studies have examined the action of VDR agonists on putative breast cancer stem or progenitor cells identified by some markers (CD44hi/CD24low and/or ADH1+) that can grow as floating, nonadherent spheres (mammospheres). In these systems, 1,25(OH)2D3 or the BXL1024 analogue reduced the population of putative CSC and the formation of mammospheres and the expression of pluripotency markers (OCT4, KL-4), Notch ligands and target genes, and genes involved in proliferation, EMT, invasion, metastasis, and chemoresistance 32,467,291 [267,268,269].
Organoids formed by cells isolated from patient-derived xenografts (not obtained directly from human biopsies but on injection and growth in mice) that acquired resistance in vitro to Trastuzumab-emtansine (T-DM1; composed of the humanized monoclonal anti-HER2 antibody Trastuzumab covalently linked to the microtubule-inhibitory agent DMI) constitute an intermediate system to the two discussed above. In this system, two vitamin D analogues (UVB1 and EM1) reduce the formation and growth of organoids [270].
5. Effects on the Immune System
1,25-(OH)2D3 is an important modulator of the immune system, as reflected by the expression of VDR by almost all types of immune cells [271,272,273]. 1,25-(OH)2D3 is an enhancer of innate immune reactions against infections and tumor cells by activating the responsive cells (macrophages, natural killer (NK) cells, and neutrophils). Conversely, and in line with its accepted anti-inflammatory action (that may contribute to the inhibition of cancers associated with chronic inflammation), 1,25-(OH)2D3 is commonly presented as a repressor of the adaptive immune reactions by deactivating antigen-presenting cells (induction of tolerogenic dendritic cells) and CD4+ type-1 helper T (Th1) response (production of interferon-γ, IL-1, IL-6, IL-12...), and by promoting the suppressive Th2 and Treg responses (production of IL-10, IL-4, IL-5, IL-13...) [273,274]. Moreover, in macrophages, 1,25-(OH)2D3 has been proposed to promote a switch from the pro-inflammatory M1 phenotype (producing IL-1β, IL-6, TNF-α, RANKL, COX) towards the anti-inflammatory protumoral M2 phenotype and to reduce the T-cell stimulatory capacity of macrophages [275,276]. This is somehow counterintuitive as it would represent a potential protumoral effect that cannot be easily attributed to a conserved evolutionary agent such as vitamin D. Some other studies discussed below have introduced putative explanations.
Since naïve T-cells express VDR at a very low level that increases only after activation of the T-cell receptor [277], the role of 1,25-(OH)2D3 may conceivably be related to the late downregulation of the activated adaptive response. This view agrees with the usual description of repressive 1,25-(OH)2D3 action in experimental settings following overstimulation of the cells, and it may constitute a safety mechanism to prevent undesirable long-lasting immune activation, potentially leading to inflammation or autoimmunity [278,279]. Concordant with this idea and the anticancer action of 1,25-(OH)2D3, a series of studies have revealed antitumor effects at the level of several types of immune cells.
Interestingly, a study in mice orthotopically implanted with breast tumors has revealed that vitamin D decreases tumor growth and increases the amount of tumor-infiltrating cytolytic CD8+ T-cells, a usual marker of antitumor response. This effect is lost in high-fat diet conditions [280]. Moreover, in pancreatic cancer, 1,25-(OH)2D3 inhibits the T-cell suppressive function of myeloid-derived suppressor cells [281].
An important mechanism of 1,25-(OH)2D3 is the inhibition of the NF-κB pathway. In turn, this causes the downregulation of multiple cytokines and their effects [282]. 1,25-(OH)2D3 inhibits NF–κB at different levels: by inactivating the p65 subunit of the NF-κB complex and upregulating the inhibitor subunit IκB. In addition, 1,25-(OH)2D3 inhibits the PG-endoperoxide synthase (PTGS-2, also known as COX-2) [283,284,285]. 1,25(OH)2D3 reduces the protumorigenic effect of PG E2 in prostate cancer cells by inhibiting COX-2 and so decreasing the levels of PG E2 and two PG receptors (EP2 and FP) [286]. Importantly, vitamin D and calcium favorably modulate the balance of expression of COX-2 and 15-hydroxyPG dehydrogenase, its physiological antagonist, in the normal-appearing colorectal mucosa of patients with colorectal adenoma [287]. Vitamin D enhances the tumoricidal activity of NK cells and macrophages [288,289]. 1,25-(OH)2D3 probably has a dual effect of stimulating the differentiation from monocytes to macrophages and their cell killing activity, including antibody-dependent cell cytotoxicity (ADCC). It may later balance these effects by promoting the M1 to M2 phenotypic switch ([279] and references therein). In addition, 1,25-(OH)2D3 enhances the susceptibility of hematological and solid cancer cells to NK cell cytotoxicity through downregulation of miR-302c and miR-520c [289].
The potentiation of ADCC of macrophages and NK cells may be a relevant antitumor action of 1,25-(OH)2D3 in clinical cases, particularly in patients treated with antibodies, of which the major mechanism of action is ADCC. Thus, several studies have shown that vitamin D deficiency impairs the macrophage and/or NK cell-mediated cytotoxicity of Rituximab (anti-CD20) in diffuse large B-cell, follicular, and Burkitt lymphoma patients [288,290,291], and of Cetuximab (anti-EGFR) in colon cancer cell lines [292]. In addition, some evidence of benefit has been observed in breast cancer patients treated with Trastuzumab (anti-HER2) and in melanoma patients treated with Bevacizumab (anti-VEGF) [290,293].
Agents that target programmed death (PD)-1 or its ligand PD-L1 immune checkpoint inhibitors (ICI) have attracted great attention in cancer therapy. Interestingly, 1,25-(OH)2D3 upregulates PD-L1 in human (but not mouse)-cultured epithelial and immune cells [294], while vitamin D treatment increases PD-1 expression in CD24+CD25+int T-cells in Crohn’s disease patients [295] and PD-L1 in epithelial and immune cells in melanoma patients [296]. These data suggest the possibility of combined treatments with VDR agonists and these ICIs, and perhaps others in development.
In conclusion, it is conceivable that 1,25-(OH)2D3 works as a general homeostatic regulator of the immune system, ensuring an appropriate global defense against challenges like tumors and infections.
6. Animal Models
Many studies on animal diet, chemical, genetic, and xenograft models (mainly for colon and breast cancer) have shown the antitumor actions of vitamin D compounds. This in vivo action is difficult to dissect and probably results from a variable combination of mechanisms in the distinct systems that were assayed, including the inhibition of tumor cell growth, EMT, invasiveness, angiogenesis, and metastasis. Importantly, as occurs in cultured cancer cells, vitamin D antitumor action is mostly independent of TP53 gene status [119,187].
7. Systemic Effects: Detoxification and Microbiome
7.1. Detoxification
The elimination of xenobiotics or the detoxification process involves chemical modification (phase I reactions: oxidation, hydrolysis, etc.) and subsequent conjugations to water-soluble molecules (phase II reactions) carried out by a large number of enzymes. 1,25-(OH)2D3 regulates some of these enzymes in the intestine and liver [297]. This may have a positive effect on the prevention of tumorigenesis and perhaps another more controversial impact on the inactivation of chemotherapeutic drugs [298].
1,25-(OH)2D3 induces CYP3A4, a major human drug-metabolizing enzyme, SULT2A, a phase II sulfotransferase, and members of the multidrug resistance-associated protein (MRP) family in colon carcinoma cells [299,300]. CYP3A4, SULT2A1, and MRP3 are involved in the elimination of lithocholic acid (LCA), a secondary bile acid LCA that induces DNA damage and inhibits DNA repair enzymes in colonic cells. Accordingly, LCA promotes colon cancer in experimental animals, and high levels of LCA have been found in colon cancer patients [301,302]. Interestingly, LCA binds weakly and activates VDR, and so it activates its own degradation [303]. Another example is enhancement by 1,25(OH)2D3 of the benzo[a]pyrene metabolism via CYP1A1 in macrophages [304].
7.2. Microbiome
Alteration of the intestinal microbiome (dysbiosis) is connected to colon cancer and possibly other neoplasias [305]. Many experimental studies in mice have shown that vitamin D deficiency promotes gut permeability, colon mucosa bacterial infiltration, and translocation of intestinal pathogens. These effects lead to changes in immune cell populations and gut inflammation, and cancer—an overall condition that is improved after vitamin D supplementation [306,307]. As bacteria lack VDR, the effect of vitamin D is mediated by the host. Importantly, genome-wide association analysis of the gut microbiome in two large cohorts of individuals identified VDR as a factor that influences the gut microbiota [308]. A conditioned medium from probiotic lactic acid bacteria showed increased expression of VDR and of its target CAMP gene encoding cathelicidin in cultured colon carcinoma cells and organoids. It protected against the inflammatory response induced by TNF-α [309]. The protective action against dysbiosis and the intestinal tumorigenesis of liganded VDR have been proposed to be at least partially mediated by the inhibition of the JAK/STAT pathway [310].
8. Discussion of Mechanistic Studies
The vast array of effects that 1,25-(OH)2D3 has in a wide variety of experimental systems of a high number of cancer types agrees with a selected evolutionary role in protection against tumoral processes. The underlying mechanisms include the control of tumor cell survival (autophagy, apoptosis) and phenotype (differentiation), and the inhibition of their proliferation, invasiveness, and metastasis; attenuation of the proliferation and phenotypic features of some CSC; modulation of the physiology of diverse non-tumoral stromal cells (fibroblasts, endothelial cells); and the regulation of several types of immune cells and responses.
Together, these effects reflect a multilevel anticancer action of vitamin D. Therefore, an appropriate vitamin D status of the organism should be maintained to minimize the risk and severe consequences of many neoplasias. Further supporting this, the toxicity of vitamin D supplementation is limited, acceptable, and clearly lower than that of current anticancer drugs and therapies. We are not aware of any other natural or synthetic compound that has such an array of antitumor activities combined with low toxicity. Doubtless, the available experimental results meet Koch’s postulate for biological causality regarding the existence of a global mechanism of action behind the association between vitamin D deficiency and high incidence and, especially, the mortality of several major cancer types found in observational and epidemiological studies. Hopefully, the further development of current and possibly, novel studies on the wide range of mechanisms of VDR agonists in a variety of biological systems will allow us to elucidate the anticancer action of vitamin D (Figure 3).
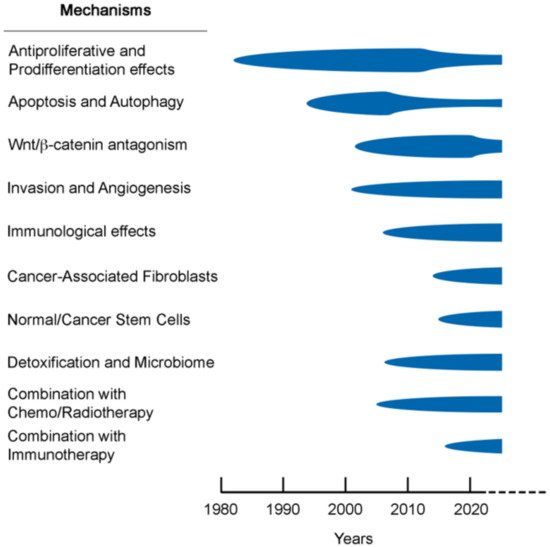
Figure 3. Time flow-chart of studies on the anticancer mechanisms of vitamin D compounds with some key references that are discussed in the text.
This entry is adapted from the peer-reviewed paper 10.3390/nu14071448
This entry is offline, you can click here to edit this entry!