Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Gliomas portray a large and heterogeneous group of CNS tumors, encompassing a wide range of low- to high-grade tumors, as defined by histological and molecular characteristics. The identification of signature mutations and other molecular abnormalities has largely impacted tumor classification, diagnosis, and therapy. Transcription factors (TFs) are master regulators of gene expression programs, which ultimately shape cell fate and homeostasis. A variety of TFs have been detected to be aberrantly expressed in brain tumors, being highly implicated in critical pathological aspects and progression of gliomas.
- transcription factors
- GLI
- E2F
- STAT3
- HIF-1/2
1. Introduction
Gliomas represent the majority (80%) of all primary malignant central nervous system (CNS) neoplasms, affecting both the brain and spinal cord. Primary CNS tumors occur more frequently in adults (29 per 10,000) than in children and adolescents; however, they are the most common types of solid tumors among pediatric cancers. According to histological similarities and cell origins, gliomas are divided to glial (astrocytomas, oligodendrogliomas, and ependymomas) and non-glial (meningiomas and medulloblastomas) tumors. The distinct types of gliomas range from grade I to IV regarding their aggressiveness and proliferative potential, with grade IV corresponding to the most malignant phenotype [1,2,3].
The pathogenesis of gliomas has been linked to several genetic alterations and deregulation of major signaling pathways. These defects include homozygous deletion of the cyclin-dependent kinase inhibitor 2A (p16) gene and complete chromosome 1p/19q deletion. They also involve mutations in tumor-suppressive genes such as Neurofibromatosis type 1 (NF1), Phosphatase and Tensin homolog (PTEN), tumor protein p53 (p53), and Retinoblastoma (RB) and in genes that are associated with metabolism and telomere length maintenance, like Isocitrate Dehydrogenase (IDH) isozyme genes and Telomerase Reverse Transcriptase (TERT), alpha-thalassemia/mental retardation, X-linked (ATRX), and Death domain Associated protein (DAXX), respectively. The signaling axis RAS/RAF/MEK is commonly dysregulated in certain types of gliomas, with a mutation in serine/threonine protein kinase BRAF where valine is substituted with glutamic acid at amino acid 600, affecting cell growth and differentiation. In addition to genetic changes, epigenetic alterations involving DNA methylation, histone modifications, and miRNAs have emerged in the last few years as important contributors to neoplastic transformation and progression due to their interplay with gene expression [3,4,5,6]. In particular, mutations affecting IDH genes result in the production of the natural metabolite α-ketoglutarate and the oncogenic byproduct, 2-hydroxyglutarate (2HG) [7]. The accumulation of 2HG leads to global DNA hypermethylation by restricting the function of TET enzymes, which are known demethylases. This swift DNA methylation pattern interferes also with the binding and activity of several transcription factors (TFs). Depending on the factors’ protein domains and corresponding motifs, the activity and binding site recognition ability of some TFs are repressed, while, in others, these features are promoted by DNA methylation [8]. In this way, epigenetic events, such as DNA methylation, may jeopardize gene expression programs.
The World Health Organization Classification, 2021 edition (hereafter, WHO 2021) on gliomas has been updated in order to encompass information on tumors’ phenotypic and genotypic profiles and improve the diagnostic and prognostic accuracy. The standard therapeutic approach for gliomas combines surgery, radiation, and chemotherapy with alkylating agents. Although, in some cases, therapy is beneficial, the most malignant types like glioblastoma (grade IV) exhibit recurrence and significant mortality. These properties are associated with a combination of biological, genetic, and signaling alterations that confer to tumor heterogeneity and diverse patient responses to therapy. Of great significance are specific cell niches inside the tumor, known as glioma stem cells (GSCs), that confer to this heterogeneity. Consequently, there is a mandatory need for the development of targeted molecular therapies and personalized therapeutic approaches [3,9].
Intracellular signaling pathways share a converging point in the nucleus where activation of specific transcription factors takes place. Gene expression is governed by the interplay between cis-regulatory elements, such as promoters, enhancers, silencers, and trans-acting factors, including TFs. Transcription factors most commonly bind directly to specific sequences on their target gene promoters but can also affect promoter activity by localizing to distal enhancer regions. These interactions evoke an increase or decrease in gene expression, affecting the protein synthesis rate and, ultimately, tailoring cellular behavior. To date, several mechanisms that lead to the deregulation of TFs have been reported in a wide range of cancers. Both indirect means, such as aberrant activity or mutations in upstream signaling molecules and cofactors, and direct means, such as deletions, amplifications, rearrangements, gain or loss-of-function point mutations in genes encoding TFs, contribute to altered function and expression of these regulatory proteins in cancer. In the aftermath of TF deregulation, a series of events depicted as hallmarks of cancer arise, which subsume uncontrolled cell proliferation, immune evasion, establishment of a stem cell-like phenotype, epithelial to mesenchymal transition (EMT), the prevention of cell death pathways, and therapeutic resistance.
2. Oncogenic Transcription Factors
Several TFs have been allocated an oncogenic role in gliomas either through deregulated expression or altered function due to fusion with other proteins, eventually affecting cell proliferation, differentiation, and apoptosis. In this section, we discuss experimental evidence on the oncogenic role of GLI, E2F, STAT, HIF, FOXM, and ATF family members and current targeting options.
2.1. GLI Transcription Factors
The Glioma-Associated Oncogene (GLI) transcription factor family consists of three members, GLI-1, -2, and -3, all of which contain conserved tandem C2H2 zinc finger domains and a consensus histidine/cysteine linker sequence between zinc fingers [9]. They recognize the GACCACCA consensus sequence on promoters of their target genes, including CDC2, hTERT, IRIS1, FOXM1, and BMI1, via the zinc finger motifs of their DNA-binding regions [11,12,13,14,15].
All members of the family are canonically activated by a multiprotein cascade involved in Hedgehog (Hh) signaling in order to regulate transcription of Hh target genes, such as PTCH1, PTCH2, and GM1. The Hh pathway plays a vital role in embryonic development, as it participates in the transmission of information to embryonic cells required for proper cell differentiation.
The regulation of the Hh signaling pathway relies on the balance between the activator and repressor forms of GLI transcription factors. Key components of the signaling cascade are the Hedgehog ligands (sonic Hh, Indian Hh, and desert Hh); Patched Receptors (PTCH1 and PTCH2); Smoothened Receptor (Smo); Suppressor of fused homolog (Sufu); protein kinase (PKA); and cyclic adenosine monophosphate (cAMP) [16]. All components of the signal transduction pathway have been detected in the primary cilia (PC) [17]. Upon absence of the Hh ligand, PTCH localizes at the PC base and suppresses the activity of Smo by inhibiting its translocation to the PC [18]. This results in the proteolytic cleavage of full-length glioma-associated oncogene (GliFL) and production of the Gli repressor (GliR) upon phosphorylation by PKA, glycogen synthase kinase-3 (GSK3), and casein kinase 1 (CK1) [19]. Subsequently, GliR binds to Hh target genes promoters, keeping them inactive. On the other hand, the binding of Hh to the PTCH1 receptor activates the signaling cascade. As a result, Smo inhibition is abrogated, and the signal gets transmitted via a cytoplasmic protein complex composed of Kif7, GliFL, and Sufu. Smo moves to the tip of PC and signals Sufu to release the Gli activator (GliA), which migrates into the nucleus and enhances gene transcription [16,20].
Deregulation of the Hh pathway, mostly activation, due to mutations at the associated genes or alterations in the expression of the signaling molecules, has been associated with developmental anomalies and various stages of carcinogenesis in different types of tumors. The key regulators of the pathway, GLIs, were first isolated from human glioblastoma cells in 1987. Since then, research advances have pointed that the expression of several Hh cascade components, such as GLI factors, PTCH, and Smo, were detected in several tumors of the nervous system, including gliomas. Their expression has also been correlated with poor prognosis of patient survival [21,22].
Among the three members of the GLI family, GLI1 is the best studied and associated to epigenetic modifications, since it has been shown to recruit histone acetyltransferase PCAF, inducing an active chromatin state on Hh target genes by increasing the H3K9 acetylation levels. GLI1, along with its truncated homolog (TGLI1), which behaves as gain-of-function GLI1, were reportedly shown to mediate angiogenesis in gliomas by targeting the VEGF, MMP2, MMP9, VEGF-C, TEM7, and proangiogenic heparanase (HPSE) genes, respectively [23,24,25,26]. The second member of the family, GLI2, was found to induce CDK6 expression by binding to its promoter, thereby mediating cell proliferation in Hh-associated medulloblastoma genetic mouse models [27]. In another study, GLI1-3 expression, along with its target genes, FOXM1 and BMI1, were present in all the tested glioma cell lines in contrast to normal brain tissue that lacked GLI1 expression. Moreover, GLI2 expression has been strongly linked to many types of glial tumors, including astrocytomas, gangliogliomas, glioblastomas, ependymomas, and oligodendrogliomas, whereas GLI1 and 3 correlated preferably with oligodendrogliomas. In addition, the GLI1 expression levels were particularly high in grade III and IV gliomas, whereas GLI2 was found overexpressed only in grade III tumors. At the same time, GLI1-2 overexpression in these tumors was suggested to impact their progression, since high-grade gliomas patients exhibited worse survival rates [28]. Finally, it is evident that GLI factors play an important role in stem cell phenotype formation by sustaining the expression of related genes, such as OCT4 or SOX2 [29].
The targeting of GLI proteins is difficult, because their binding domains constitute a limiting parameter for the design of small repressive molecules against them [30]. Nevertheless, GLI antagonists GANT-61 and -58 and Arsenic Trioxide (As2O3) have been developed but, to our knowledge, have not been tested in gliomas yet [31,32,33]. Some compounds targeting the Hh pathway show promise in the treatment of Medulloblastoma (MB) by overcoming the frequent phenomenon of mutation-driven drug resistance that SMO antagonists face. These compounds are effective towards both the Hedgehog pathway and the bromodomain-containing protein 4 (BRD4). This function leads to an indirect restriction of GLI activity, since BRD4 has been reported to interact with GLI1 and GLI2 promoter regions through its bromodomains and affect, in a certain amount, their expression. Liu et al. optimized the structure of 4-Aryl-1,6-dihydro-7H-pyrrolo[2,3-c]pyridin-7-one 2 (ABBV-075), among other BRD4 nonspecific inhibitors that also exhibited Hh pathway restrictive potential. Consequently, they generated a derivative compound 25 by fusing a fluoro substituent at the C3 position of the pyrrole core and compound 35, with 4-methylcyclohexyl amino ousting the phenylether motif. Both molecules were shown to be efficient GLI inhibitors, while compound 25 was further shown to abrogate tumor growth in vivo [30].
2.2. E2F Transcription Factors
The cyclin-dependent kinase (CDK)-Rb-E2F axis directs cell cycle progression, overseeing the timing and integrity of genetic material replication. Critical regulators of the pathway are members of the E2F transcription factor family. This family can be divided into three groups according to the structure and function of its members: activators (E2F1–3A), canonical repressors (E2F3B–6), and atypical repressors (E2F7 and E2F8) [34]. The levels of activator proteins peak during the G1-S phase transition, whereas atypical repressor levels peak in the succeeding S phase. Canonical repressors are constitutively expressed during all the phases of the cell cycle [35].
E2F factors contain a highly resembling winged helix DNA-binding domain (DBD) and share the ability to recognize and bind to the classic E2F consensus sequence TTCCCGCC (or slight variations of it) of their target gene promoters [36]. The DNA-binding ability of E2F1–6 transcription factors also depends on a dimerization (DIM) domain, which is composed of a leucine zipper (LZ) and a marked box (MB) domain [37]. To activate transcription, canonical E2Fs need to form a complex with a member of the transcription factor dimerization partner family (TFDP1, TFDP2 and TFDP3). E2F1–5 factors also carry a transactivation domain that binds pocket proteins (RB, p107, and p130) [38,39]. Upon RB presence, E2F activators are unable to promote cell cycle progression. On the contrary, E2F7 and E2F8, containing two tandem E2F DBDs, interact to form a single DNA-binding surface that recognizes the E2F consensus sequence independently of TFDP proteins (Figure 1) [40].
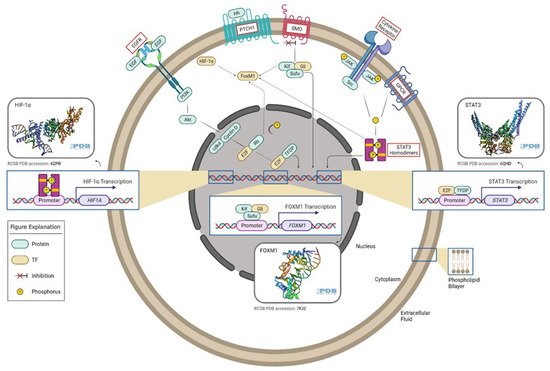
Figure 1. Oncogenic transcription factors and associated signaling pathways in gliomas. E2F TF is a downstream factor of the EGFR/PI3K/Akt pathway. The Rb tumor suppressor protein (pRb) binds to the E2F1 transcription factor, preventing it from interacting with the cell’s transcriptional machinery. When pRb gets phosphorylated, it detaches from E2F. E2F (along with its binding partner, TFDP) mediates the transactivation of E2F1 target genes, such as STAT3. GLI TF is a downstream effector of the Hedgehog pathway. In the absence of Hh, PTCH acts to prevent high expression and activity of SMO. GLI TFs function in a complex with Kif7 and Sufu, translocate to the nucleus, and induce the expression of target genes such as FoxM1. STAT3 TFs can be activated by several signals, which involve G-protein-coupled (GPCR) and cytokine receptors. Phosphorylated STAT3 homodimers regulate HIF-1α expression. FoxM1 can be activated by STAT3, GLI, HIF-1α, and E2F TFs. This figure was created with the tools provided by BioRender.com, accessed on 22 February 2022.
E2F factors and associated genes exhibit altered expressions in gliomas, according to a variety of studies. All E2Fs (except for E2F3 and E2F5) are highly expressed in high-grade gliomas (HGG) and linked to grade progression, indicating an adverse outcome [41]. Nonetheless, Li et al. portrayed a mechanism for glioma progression in their study, which involved the upregulation of E2F3. Overall, they demonstrated through several functional assays, MS2-RIP, and siRNA transfections that the lncRNA SNHG5 acts as an oncogenic factor in gliomas by competitively engaging (sponging) miR-205 and suppressing its function. Therefore, miR-205 is unable to bind its target sequences on E2F3 3′UTR, resulting in the upregulation of E2F3 expression. They also showed that this mechanism drives glioma cell migration and invasion and increases glucose uptake in vitro, while its inhibition curtails tumor growth in vivo [42].
Regarding related genes, the upregulation of genes encoding DP family members has been reported alongside a significant rise in E2F1 mRNA levels [43]. Moreover, Zhi et al. unveiled a potential mechanism by which ECT2 facilitates glioma cell proliferation both in vitro and in vivo. In their study, the ECT2 expression levels were increased in glioma cell lines and tissues compared to normal brain tissue and human astrocytes (NHAs) and correlated with the tumor grade. In summary, they suggested a pathway where ECT2 regulates the expression of PMSD14 deubiquitinase, which, in turn, stabilizes the E2F1 factors and prevents its degradation by proteasome machinery, resulting in PTTG1 upregulation. Keeping in mind that PTTG1 can mediate glioma cell proliferation, the signaling cascade ECT2/PMSD14/E2F1/PPT11G could be potentially targeted as a therapeutic approach [44,45].
In the recent study of Yu et al., E2F8 expression was found augmented in gliomas compared to normal brain tissues, especially in all four glioblastoma (GBM) subtypes (classical, mesenchymal, neural, and pro-neural), and associated with poor outcome regarding patients’ survival. Further investigation of E2F8 role in GBM revealed an attenuated proliferation of GBM cells and prolonged survival of animal models upon E2F8 gene silencing. In addition, bioinformatic analysis pointed out a tight association of E2F8 expression with aggressive cell cycle induction; DNA repair process; and key signaling pathways (STAT3, TGFRβ, and WNT). Moreover, the results from a correlation expression analysis and latter ChIP-PCR suggested E2F8 as a key candidate for CHEK1 transcriptional activity regulation in GBM tumor cells. Collectively, these data demonstrate that E2F8 plays a pivotal role in cell proliferation, tumor formation, and multiple oncogenic processes in GBM [46].
Yang et al. investigated E2F7 role and function in gliomas and observed an upregulation of this TF in GBM patients, which was associated with poor overall survival. In vitro functional studies and in vivo model experiments revealed that E2F7 induced cell proliferation, cell cycle progression, and metastasis featuring tumorigenic abilities. Moreover, functional studies on E2F7 promotion of transcription and its participation in epigenetic mechanisms revealed that E2F7 binds to EZH2 promoter, activating its transcription and increasing the H3K27me3 levels. Subsequently, EZH2 recruited H3K27me3 to PTEN’s promoter, inhibiting its expression and turning on the AKT/mTOR signaling pathway. Seemingly, E2F7 tumorigenic properties rely on the EZH2-mediated PTEN/AKT/mTOR pathway in GBM [40,47]. In addition, Lu et al. uncovered the role of lncRNA SNHG12, which has been found overexpressed in GBM cell lines and tissues as a mediator of cell proliferation and resistance to treatment with temozolomide (TMZ) in GBM. The overexpression of SNHG12 is attributed to a decline in DNA methylation at its promoter, which enables the engagement of SP1 transcription factor and, ultimately, transcriptional induction. Furthermore, the study demonstrated that miR-129-5p gets sponged by SNHG12, and its downregulation was involved in the promotion of TMZ resistance. As an outcome, MAPK1 and E2F7, which carry binding sites for miR-129-5p at their 3′UTRs, were detected upregulated in TMZ-resistant GBM cells. Although the knockdown of both genes altered the resistant phenotype and cell proliferation rate, the E2F7 factor was mainly linked to G1/S transition, while MAPK1 is implicated in both G1/S transition and cell apoptosis with regards to TMZ treatment [48].
2.3. STAT3 Transcription Factor
Signal Transducer and Activator of Transcription 3 (STAT3) belongs in the family of STAT proteins composed of signal transducers and transcription regulators. The family encompasses seven members (STAT1, 2, 3, 4, 5A, 5B, and 6) that are encoded by different genes and exhibiting different functions but sharing a common structure [49]. The protein structure consists of six functional domains: an N-terminal, a coiled-coil (CC), a DBD, a linker sequence, Src Homology 2 (SH2), and finally, a transactivation domain (TAD). Of great significance are a tyrosine residue at amino acid position 705 (Tyr705) located in the SH2 domain and a serine phosphorylation site at residue 727 (Ser727) within the C-terminal domain, both involved in STAT activation [50].
The gene encoding STAT3, the third member of the family, resides at the 17q21.31 genomic region. To date, two isoforms of STAT3, the full-length STAT3α (770aa) and the truncated STAT3β (722aa), have been identified. They arise from alternative splicing and proteolytic cleavage processes. Interestingly, the truncated forms of STAT proteins, such as STAT3β, act mainly as dominant-negative of the corresponding full-length proteins [51,52].
STAT3 activation can be induced by extrinsic and intrinsic stimuli associated with cytokine signaling; some plasma membrane receptors (EGFR and PDGFR); and cytoplasmic kinases (Src family, BMX, and Bcr-Abl fusion protein), respectively (Figure 1). The phosphorylation of Y705 residue at the carboxyterminal is the most frequent of STAT3 modifications and is considered as its canonical activation marker. This can be achieved either by recruitment of JAK kinases to the receptor’s cytoplasmic tail or directly by specific cytoplasmic kinases. Additional posttranslational modifications of STAT3 that lead to its activation include phosphorylation at Ser727, acetylation (at lysine residues K49 and K87), and methylation (at lysine residue K140) [53,54,55]. STAT3 can be regulated by several mechanisms, including a synthesis/degradation cycle dependent on proteasome function and an activation/inactivation loop, mostly linked to its phosphorylated/dephosphorylated state (Figure 1).
Upon phosphorylation, STAT3 undergoes dimerization via reciprocal interactions with SH2 domains that bind to phosphotyrosine. Thereafter, the formed active homo- and heterodimers can translocate to the nucleus and stimulate transcription through recognition of the small palindromic consensus sequence TTCN2-4GAA that defines GAS elements within target gene promoters [48].
Of all members of the family, STAT3 is most frequently implicated in various types of cancers [53]. The deregulation and constant activation of STAT3 in gliomas is considered to result from an aberrant signal from upstream regulators, since no gain-of-function mutation of this molecule has been identified yet. On the one hand, this speculation concerns gain-of-function mutations or enhanced activation of an upstream activator, while on the other hand, it entails loss-of-function mutations or reduced activation of an upstream repressor [49]. These further lead to alterations in signaling pathways mediated by receptor-associated tyrosine kinase activities where growth factor receptors and cytokines are key components and upregulation of protein serine/threonine kinases.
Abnormally redundant signaling that occurs from gene amplifications and/or rearrangements of EGFR gives rise to the formation of a truncated variant (EGFRvIII) or of the fusion mutant EGFR-SEPT14, resulting in a hyperactive pSTAT3-Y705 molecule. Puram et al. demonstrated that STAT3 promotes transcriptional regulation of inducible nitric oxide synthase (iNOS) in GBs, which specifically carry the activated EGFRIII variant. STAT3 was associated with tumor progression and invasive aptitude [56,57]. Another study focusing on TGF-β, a multifunctional polypeptide growth factor, showed that TGF-β-related glioma cells invasion required phosphorylation of STAT3 at the Y705 residue via IFITM3-STAT3 axis [58]. Moreover, cytokines IL-6 and OSM have been detected overexpressed in gliomas. Both cytokines induce STAT3 phosphorylation at Tyr Y705 through the hexameric receptor complex IL-6Rα. In particular, OSM contributes to the aggressiveness of the mesenchymal subtype and has the ability to activate STAT3 expression by forming a complex with EGFRvIII, which, at the end results, in its overexpression.
It is also notable that activation of STAT3 in GBM stem-like cells has been linked to the activity of non-tyrosine kinases, like the bone marrow and X-linked (BMX) nonreceptor tyrosine kinase [49,59,60]. Serine/threonine kinases mediate STAT3 phosphorylation at serine 727 residue. For instance, PKCε (Protein Kinase C epsilon) overexpression in human anaplastic astrocytoma and GBM cases seems to consort constitutive activation of STAT3 through serine 727 phosphorylation [61,62].
At the same time, deregulation of STAT3 expression may also refer to the deficiency of upstream repression regulators, such as PIAS3, SOCS, and PTPRD. For instance, a reduced expression of PIAS3 accompanied by elevated pSTAT3-Y705 levels has been observed in GBM, in contrast with normal brain tissues [48,63].
Regardless of the mechanism behind its activation, STAT3 undoubtedly possesses a crucial part in the pathogenesis of gliomas, the proliferation and migration of glioma cells, while contributing to the stem-like phenotype, angiogenesis, and immune suppression. Regarding cell survival and proliferation, several in vivo and in vitro approaches that focus on STAT3 inhibition have demonstrated a mitigated accumulation of antiapoptotic factors, such as Survivin, Bcl-2, Bcl-Xl, and Mcl-1, and a concomitant attenuated expression of cell cycle regulators, like c-myc, cyclin E, and cyclin D1 [64,65]. Besides cell proliferation, STAT3 has been linked to migration and invasion of glioma cells. According to some studies, inhibition of STAT3 led to reduced production of matrix metalloproteinases (MMP2 and MMP9) and was associated with genes that account for EMT, namely Snail [66]. In addition, STAT function is associated with p65-NF-κB and nuclear factor I-X3, resulting in the upregulation of ICAM-1 and YK1-40, respectively, fueling the migration and invasion abilities of glioma cells [67].
Moreover, STAT3 activation due to plasma membrane and cytokine stimuli has been shown to induce immune tolerance. Its activation attenuates differentiation, maturation, and functions of dendritic cells; disrupts T-cell proliferation; and promotes T-cell anergy and immunosuppressive microglia [4,68,69].
Moreover, STAT3 transcription factor inhibition is considered as a promising approach for glioma treatment, mostly in GBM cases since it drives pro-neural–mesenchymal transition and is implicated in the aggressiveness and stemness of glioma tumors. The study of Tan et al. distinguished two subgroups based on a transcriptomic signature associated with the STAT3 pathway that could help to predict the patients’ response to therapy with STAT3 inhibitors. STAT3high defined a patient cohort enriched in the mesenchymal and classical molecular subtypes with non 1p/19q codeletion and IDH-WT status, describing highly aggressive and recurrent gliomas. On the contrary, STAT3low tumors are comprised mostly of low-grade gliomas (LGGs) and the pro-neural molecular subtype with enrichment of 1p/19q codeletion and IDH-mutant status, presenting tumors of better prognosis and responsiveness to current chemotherapy. They demonstrated that dual inhibition of IGF-1R with NT157 and STAT3 with AZD1480 and/or Linsitinib sensitizes STAT3-low cells and improves survival. The latter also functions synergistically with the TMZ standard treatment [70]. Likewise, JSI-124 (cucurbitacin I), a natural chemical compound, has been shown to suppress the expression of VEGF and blocked the phosphorylation of JAK2 in a dose-dependent manner. Thus, the antiangiogenic effects of JSI-124 might occur through VEGFR2/STAT3 (Ser727) inhibition [71]. Additionally, JSI-124 was shown to sensitize glioma cells to DNA-alkylating agents TMZ and cisplatin [72]. Other STAT3 pharmacological inhibitors, such as AG490, WP1066, LLL3, and Gefitinib, have also exhibited potential therapeutic benefits [73,74]. Especially WP1066, which explicitly targets glioma cells leaving intact normal astrocytes, can shift immune tolerance in glioma patients by inducing the production of costimulatory factors in macrophages that infiltrate glioma tumors and cytokines that trigger effector T cells [75]. Additionally, Ibrutinib (PCI-32765), which is an approved small molecule for the treatment of mantle cell lymphoma and chronic lymphocytic leukemia, has been shown to target BMX in GSCs and alleviate tumor expansion in GSC-derived orthotopic xenografts. Therefore, Ibrutinib constitutes an attractive option for the indirect inhibition of STAT3 hyperactivation in GBM [76]. Kadiyala et al. designed albumin-based nanoparticles (NPs) bearing the tumor-penetrating peptide iRGD to successfully infiltrate the blood–brain barrier, deliver specific small-interfering RNA (siRNA), and silence STAT3 expression in GBM tumors. This is a very promising approach, since the NPs induced prolonged survival in synergy with ionizing radiation (IR) treatment and immunological memory against GBM recurrence in mice [77].
2.4. HIF Transcription Factors
The maintenance of oxygen homeostasis is crucial especially for organisms like metazoans, which rely mostly on aerobic energy production. Hypoxia-inducible transcription factors (HIFs) are key regulators of gene expression in hypoxic conditions featuring reduced oxygen levels. Genes that are activated upon oxygen reduction are those implicated in mitochondrial function, energy metabolism, oxygen binding, and delivery, as well as hematopoiesis [78,79]. HIFs are also responsible for the regulation of VEFG expression and may be involved in the formation of the endothelium that gives rise to the blood–brain barrier.
Structurally, HIFs is composed of two subunits that can form a functional heterodimer in order to regulate transcription. Three paralogs of the HIF-α (HIF-1α, HIF-2α/EPAS, and HIF-3α) and two paralogs of the HIFβ (ARNT and ARNT2) subunit have been detected [79]. The α-subunits are oxygen-responsive cytoplasmic proteins, whereas β-subunits are nuclear proteins expressed in a constant rate. The proteins of this family are defined by the existence of an N-terminal bHLH (basic helix-hoop-helix) DNA-binding domain upstream of two per-ARNT-Sim (PAS) domains [80]. The α-subunits may also contain an oxygen-dependent degradation domain (ODDD) serving as an inhibitory element and an N-terminal translocation domain (NTAD). In addition to the previous domains, HIF-1α and HIF-2α contain a C-terminal transactivation domain (CTAD) [81].
Under normal oxygen concentration, HIF α-subunits undergo degradation through hydroxylation by prolyl hydroxylase domain protein (PHD) and polyubiquination by Von Hippel-Lindau (VHL), assisted by the E3 ligase. The modified α-subunit is then degraded at the proteasome. On the contrary, during hypoxia, the activity of PHD is reduced resulting in the cytoplasmic HIF-α stabilization, accumulation, and translocation to the nucleus. There, the α-subunits dimerize with either one of the β-subunits assisted by bHLH and PAS domains. For HIFs to regulate gene transcription, bHLH domains must come into contact with the core nucleotides of HIF-responsive elements (HRE) within gene promoter regions and mediate their binding (Figure 1) [82].
Hypoxic conditions are considered as a common outcome of tumor progression and development among different cancer types, because cancer cells proliferate rapidly outgrowing the tumor’s blood supply. GBM tumors, probably because of their aggressive nature, are very keen to develop perivascular hypoxia. This is supported by immunohistochemistry that identifies HIF-2α expression in GBMs [83].
HIF-2α protein is closely linked to the stem phenotype of glioma cells, which is essential for tumor recurrence and resistance to therapy. The transcription factor is selectively upregulated in GSCs but absent in normal progenitor cells [84,85]. Although the mechanisms that underlie its upregulation are not completely understood, a recently identified gain-of-function missense mutation in the oxygen-dependent degradation domain may be a possible explanation, since it prevents its degradation [83]. HIF-2α expression has been linked to transmembrane CD44 glycoprotein produced by stem cells in the perivascular niche of GBMs. Functional studies, employing knockdown of the factor in GSCs, demonstrated decreased tumor sphere formation, reduced GSC-mediated angiogenesis (in vitro), induction of cell apoptosis, and repression of GSC oncogenes transcription. According to evidence from in vivo experiments, the knockdown of HIF-2α in glioma xenograft models increased survival and stalled the appearance of neurological impairment. Concomitantly, upon CD44 intracellular domain inhibition, a downregulation of HIF-2α and a containment of hypoxia-induced glioma stemness were observed [86,87,88]. Moreover, regarding patients’ survival, clinical trials, and REMBRANT database, an inverse correlation with HIF-2α expression was supported. In addition, an overexpression of HIF-2α was witnessed in several chemo-resistant cell lines [85,89].
HIFs also seem to drive the metabolic reprogramming of branched-chain amino acids (BCAAs) in GBM in response to hypoxia. BCAAs, including leucine, isoleucine, and valine, are transported to the cytosol by members of the L-type amino acid transporters family (LAT1-4) and catabolized by branched-chain aminotransferases BCAT1 and BCAT2. In GBM cells, HIF-1 and HIF-2 induce LAT1 upregulation. In particular, HIF-1α solely mediates BCAT1 transcription in GBM cells, notwithstanding that both proteins are able to bind directly to the HRE at the first intron of the BCAT1 human gene. Additional evidence of HIF-mediated reprogramming of BCAA metabolism relies on the fact that knockout of HIF1A and HIF2A significantly reduced glutamate labeling of BCAAs in GBM cells in hypoxic conditions. Altogether, HIF family is important for cell homeostasis and its members have been risen as possible mediators of tumor progression [90].
Regarding the effects of hypoxic conditions in transcription of certain genes that promote the malignant properties of gliomas and neoangiogenesis, inhibition of HIF TFs and their signaling pathway components has caught the attention of the research community as a possible molecular therapeutic target. Specifically, Acriflavine (ACF), an FDA-approved small molecule, can be administered locally in the brain by penetrating the blood–brain barrier via biodegradable polymers and drive the apoptosis of glioma cells. The pathway leading to apoptosis involves the reduction of HIF-1α and its target genes (PGK-1 and VEGF) expression, suggesting that HIF pathway inhibition drives ACF-mediated glioma cell death. These findings are of immense importance, since ACF results in almost 100% long-term survival, as confirmed by MRI and histological analysis [91]. In a comparable way, cyclic peptide inhibitor cyclo-CLLFVY and PT2385 or PT2977 interfere with the HIF-α/HIF-β dimerization process by interacting with the PAS domains of HIF-1α and HIF-2α, respectively [92]. Additionally, the topoisomerase inhibitor Topotecan attenuates tumor growth and angiogenesis through the inhibition of HIF-1α and its target genes expression in GBM in vivo models [93]. In accordance, a combinational treatment with Topotecan and Bevacizumab, a humanized monoclonal antibody against VEGF, has been reported to exert antiproliferative function towards glioma cells due to HIF-1α activity reduction [94]. Another novel small molecule, 103D5R, decreases HIF-1α expression, inhibits the transcription of HIF-1α target genes and prevents angiogenesis and metabolic adaptation in gliomas [95]. Moreover, the natural polyphenolic compound Vitexin was shown to repress HIF-1α expression. This flavonoid has been shown to comply with Hyperbaric oxygen (HBO) in increasing the sensitivity of glioma tumors to radiotherapy in mice [96]. In addition, Borneolum Syntheticum, commonly known as Borneol, is a bicyclic monoterpenoid reported to mediate apoptotic processes in glioma cells in vitro by overseeing HIF1α expression [97].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073720
This entry is offline, you can click here to edit this entry!